Methylation of benzene by methanol: single-site kinetics over H-ZSM-5 and H-beta zeolite catalysts
Abstract
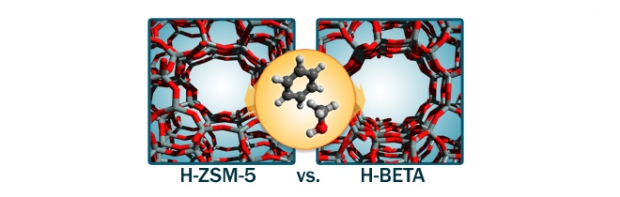
Benzenemethylation by methanol is studied on acidic zeolitesH-ZSM-5 (MFI) and H-beta (BEA) to investigate the influence of the catalyst topology on the reaction rate. Experimental kinetic measurements at 350 °C using extremely high feed rates to suppress side reactions show that methylation occurs considerably faster on H-ZSM-5 than on H-beta. Theoretical rate constants, obtained from first-principles simulations on extended zeolite clusters, reproduce a higher methylation rate on H-ZSM-5 and provide additional insight into the various molecular effects that contribute to the overall differences between the two catalysts. The calculations indicate this higher methylation rate is primarily due to an optimal confinement of the reacting species in the medium pore material. Co-adsorption of methanol and benzene is energetically favored in H-ZSM-5 compared with H-beta, to the extent that the stabilizing host–guest interactions outweigh the greater entropy loss upon benzene adsorption in H-ZSM-5 vs. in H-beta.
 Open Access version available at UGent repository
Open Access version available at UGent repository