L. Vanduyfhuys
Reaching quantum accuracy in predicting adsorption properties for ethane/ethene in ZIF-8 at the low pressure regime

Journal of Chemical Theory and Computation
20, 12, 5225-5240
2024
A1
Abstract
Nanoporous materials in the form of metal−organicframeworks such as zeolitic imidazolate framework-8 (ZIF-8) arepromising membrane materials for the separation of hydrocarbonmixtures. To compute the adsorption isotherms in suchadsorbents, grand canonical Monte Carlo simulations have provento be very useful. The quality of these isotherms depends on theaccuracy of adsorbate−adsorbent interactions, which are mostlydescribed using force fields owing to their low computational cost.However, force field predictions of adsorption uptake often showdiscrepancies from experiments at low pressures, providing theneed for methods that are more accurate. Hence, in this work, wepropose and validate two novel methodologies for the ZIF-8/ethane and ethene systems; a benchmarking methodology toevaluate the performance of any given force field in describing adsorption in the low-pressure regime and a refinement procedure torescale the parameters of a force field to better describe the host−guest interactions and provide for simulation isotherms with closeagreement to experimental isotherms. Both methodologies were developed based on a reference Henry coefficient, computed withthe PBE-MBD functional using the importance sampling technique. The force field rankings predicted by the benchmarkingmethodology involve the comparison of force field derived Henry coefficients with the reference Henry coefficients and ranking theforce fields based on the disparities between these Henry coefficients. The ranking from this methodology matches the rankingsmade based on uptake disparities by comparing force field derived simulation isotherms to experimental isotherms in the low-pressure regime. The force field rescaling methodology was proven to refine even the worst performing force field in UFF/TraPPE.The uptake disparities of UFF/TraPPE improved from 197% and 194% to 11% and 21% for ethane and ethene, respectively. Theproposed methodology is applicable to predict adsorption across nanoporous materials and allows for rescaled force fields to reachquantum accuracy without the need for experimental input.
High-Throughput Screening of Covalent Organic Frameworks for Carbon Capture Using Machine Learning
Chemistry of Materials
36, 9, 4315-4330
2024
A1
Abstract
Postcombustion carbon capture provides a high-potential pathway to reduce anthropogenic CO2 emissions in the short term. In this respect, nanoporous materials, such as covalent organic frameworks (COFs), offer a promising platform as adsorbent beds. However, due to the modular nature of COFs, an almost unlimited number of structures can possibly be synthesized. To efficiently identify promising materials and reveal performance trends within the COF material space, we present a computational high-throughput screening of 268,687 COFs for their ability to efficiently and selectively separate CO2 from the flue gas of power plants using a pressure swing adsorption process. Furthermore, we demonstrate that our screening can be significantly accelerated using machine learning to identify a set of promising materials. These are subsequently characterized with high accuracy, taking into account the effects of competitive adsorption and coadsorption. Our screening reveals that imide, (keto)enamine, and (acyl)hydrazone COFs are particularly interesting for carbon capture. Additionally, the best-performing materials are 3D COFs possessing strong CO2 adsorption sites between aromatic rings at opposite sides of pores with a diameter of 1.0 nm. In 2D COFs, a significant influence of the framework chemistry, such as imide linkages or fluoro groups, is observed. Our design rules can guide experimental researchers to construct high-performing COFs for CO2 capture.
Gold Open Access
OGRe: Optimal grid refinement protocol for accurate free energy surfaces and its application to proton hopping in zeolites and 2D COF stacking
Journal of Chemical Theory and Computation
19, 24, 9032-9048
2024
A1
Abstract
While free energy surfaces are the crux of our understanding in many chemical and biological processes, their accuracy is generally unknown. Moreover, many developments to improve their accuracy are often complicated, impeding their general use. Luckily, several tools and guidelines are already in place to identify these shortcomings, but they are typically lacking in flexibility or fail to systematically determine how to improve the accuracy of the free energy calculation. To overcome these limitations, this work introduces OGRe--a python package for optimal grid refinement in an arbitrary number of dimensions. OGRe is based on three metrics which gauge the confinement, consistency, and overlap of each simulation in a series of umbrella sampling (US) simulations, an enhanced sampling technique ubiquitously adopted to construct free energy surfaces for hindered processes. As these three metrics are fundamentally linked to the accuracy of the weighted histogram analysis method, adopted to generate free energy surfaces from US simulations, they facilitate a systematic construction of accurate free energy profiles, where each metric is driven by a specific umbrella parameter. This allows for the derivation of a consistent and optimal collection of umbrellas for each simulation, largely independent of the initial values, thereby dramatically increasing the ease-of-use towards accurate free energy surfaces. As such, OGRe is particularly suited to determined complex free energy surfaces, with large activation barriers and shallow minima, which underpin many physical and chemical transformations, and hence to further our fundamental understanding of these processes.
Gold Open Access
DFT-Quality Adsorption Simulations in Metal–Organic Frameworks Enabled by Machine Learning Potentials
Journal of Chemical Theory and Computation (JCTC)
19, 18, 6313-6325
2023
A1
Abstract
Nanoporous materials such as metal–organic frameworks (MOFs) have been extensively studied for their potential for adsorption and separation applications. In this respect, grand canonical Monte Carlo (GCMC) simulations have become a well-established tool for computational screenings of the adsorption properties of large sets of MOFs. However, their reliance on empirical force field potentials has limited the accuracy with which this tool can be applied to MOFs with challenging chemical environments such as open-metal sites. On the other hand, density-functional theory (DFT) is too computationally demanding to be routinely employed in GCMC simulations due to the excessive number of required function evaluations. Therefore, we propose in this paper a protocol for training machine learning potentials (MLPs) on a limited set of DFT intermolecular interaction energies (and forces) of CO2 in ZIF-8 and the open-metal site containing Mg-MOF-74, and use the MLPs to derive adsorption isotherms from first principles. We make use of the equivariant NequIP model which has demonstrated excellent data efficiency, and as such an error on the interaction energies below 0.2 kJ mol–1 per adsorbate in ZIF-8 was attained. Its use in GCMC simulations results in highly accurate adsorption isotherms and heats of adsorption. For Mg-MOF-74, a large dependence of the obtained results on the used dispersion correction was observed, where PBE-MBD performs the best. Lastly, to test the transferability of the MLP trained on ZIF-8, it was applied to ZIF-3, ZIF-4, and ZIF-6, which resulted in large deviations in the predicted adsorption isotherms and heats of adsorption. Only when explicitly training on data for all ZIFs, accurate adsorption properties were obtained. As the proposed methodology is widely applicable to guest adsorption in nanoporous materials, it opens up the possibility for training general-purpose MLPs to perform highly accurate investigations of guest adsorption.
Operando modeling of zeolite catalyzed reactions using first principle molecular dynamics simulations
ACS Catalysis
13, 17, 11455-11493
2023
A1
Abstract
Within this Perspective, we critically reflect on the role of first-principles molecular dynamics (MD) simulations in unraveling the catalytic function within zeolites under operating conditions. First-principles MD simulations refer to methods where the dynamics of the nuclei is followed in time by integrating the Newtonian equations of motion on a potential energy surface that is determined by solving the quantum-mechanical many-body problem for the electrons. Catalytic solids used in industrial applications show an intriguing high degree of complexity, with phenomena taking place at a broad range of length and time scales. Additionally, the state and function of a catalyst critically depend on the operating conditions, such as temperature, moisture, presence of water, etc. Herein we show by means of a series of exemplary cases how first-principles MD simulations are instrumental to unravel the catalyst complexity at the molecular scale. Examples show how the nature of reactive species at higher catalytic temperatures may drastically change compared to species at lower temperatures and how the nature of active sites may dynamically change upon exposure to water. To simulate rare events, first-principles MD simulations need to be used in combination with enhanced sampling techniques to efficiently sample low-probability regions of phase space. Using these techniques, it is shown how competitive pathways at operating conditions can be discovered and how broad transition state regions can be explored. Interestingly, such simulations can also be used to study hindered diffusion under operating conditions. The cases shown clearly illustrate how first-principles MD simulations reveal insights into the catalytic function at operating conditions, which could not be discovered using static or local approaches where only a few points are considered on the potential energy surface (PES). Despite these advantages, some major hurdles still exist to fully integrate first-principles MD methods in a standard computational catalytic workflow or to use the output of MD simulations as input for multiple length/time scale methods that aim to bridge to the reactor scale. First of all, methods are needed that allow us to evaluate the interatomic forces with quantum-mechanical accuracy, albeit at a much lower computational cost compared to currently used density functional theory (DFT) methods. The use of DFT limits the currently attainable length/time scales to hundreds of picoseconds and a few nanometers, which are much smaller than realistic catalyst particle dimensions and time scales encountered in the catalysis process. One solution could be to construct machine learning potentials (MLPs), where a numerical potential is derived from underlying quantum-mechanical data, which could be used in subsequent MD simulations. As such, much longer length and time scales could be reached; however, quite some research is still necessary to construct MLPs for the complex systems encountered in industrially used catalysts. Second, most currently used enhanced sampling techniques in catalysis make use of collective variables (CVs), which are mostly determined based on chemical intuition. To explore complex reactive networks with MD simulations, methods are needed that allow the automatic discovery of CVs or methods that do not rely on a priori definition of CVs. Recently, various data-driven methods have been proposed, which could be explored for complex catalytic systems. Lastly, first-principles MD methods are currently mostly used to investigate local reactive events. We hope that with the rise of data-driven methods and more efficient methods to describe the PES, first-principles MD methods will in the future also be able to describe longer length/time scale processes in catalysis. This might lead to a consistent dynamic description of all steps─diffusion, adsorption, and reaction─as they take place at the catalyst particle level.
Undercoordinated confined water in Brønsted acidic zeolites speeds up the O-activated demethylation of guaiacol in hot-pressurized water
Mechanistic characterization of zeolite-catalyzed aromatic electrophilic substitution at realistic operating conditions
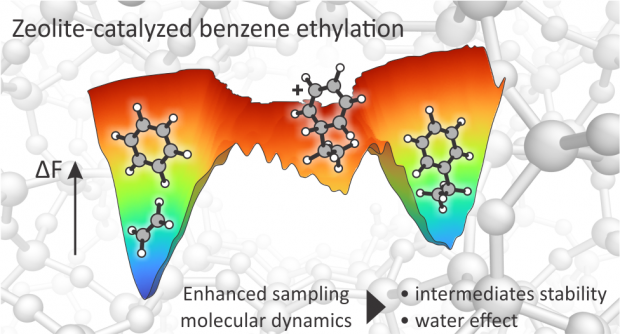
JACS Au (Journal of the American Chemical Society)
2, 2, 502-514
2022
A1
Abstract
Zeolite-catalyzed benzene ethylation is an important industrial reaction, as it is the first step in the production of styrene for polymer manufacturing. Furthermore, it is a prototypical example of aromatic electrophilic substitution, a key reaction in the synthesis of many bulk and fine chemicals. Despite extensive research, the reaction mechanism and the nature of elusive intermediates at realistic operating conditions is not properly understood. More in detail, the existence of the elusive arenium ion (better known as Wheland complex) formed upon electrophilic attack on the aromatic ring is still a matter of debate. Temperature effects and the presence of protic guest molecules such as water are expected to impact the reaction mechanism and lifetime of the reaction intermediates. Herein, we used enhanced sampling ab initio molecular dynamics simulations to investigate the complete mechanism of benzene ethylation with ethene and ethanol in the H-ZSM-5 zeolite. We show that both the stepwise and concerted mechanisms are active at reaction conditions and that the Wheland intermediate spontaneously appears as a shallow minimum in the free energy surface after the electrophilic attack on the benzene ring. Addition of water enhances the protonation kinetics by about 1 order of magnitude at coverages of one water molecule per Brønsted acidic site. In the fully solvated regime, an overstabilization of the BAS as hydronium ion occurs and the rate enhancement disappears. The obtained results give critical atomistic insights in the role of water to selectively tune the kinetics of protonation reactions in zeolites.
Gold Open Access
Crystals springing into action: metal-organic framework CUK-1 as a pressure-driven molecular spring dagger
Chemical Science
12, 5682-5687
2021
A1
Abstract
Mercury porosimetry and in situ high pressure single crystal X-ray diffraction revealed the wine-rack CUK-1 MOF as a unique crystalline material capable of a fully reversible mechanical pressure-triggered structural contraction. The near-absence of hysteresis upon cycling exhibited by this robust MOF, akin to an ideal molecular spring, is associated with a constant work energy storage capacity of 40 J/gr. Molecular simulations were further deployed to uncover the free-energy landscape behind this unprecedented pressure-responsive phenomenon in the area of compliant hybrid porous materials. This discovery is of utmost importance from the perspective of instant energy storage and delivery.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGreen Open Access