Abstract
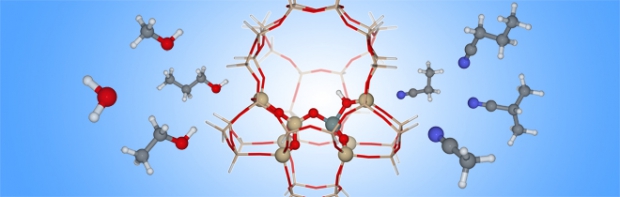
Since many industrially important processes start with the adsorption of guest molecules inside the pores of an acidic zeolite catalyst, a proper estimate of the adsorption enthalpy is of paramount importance. In this contribution, we report ab initio calculations on the adsorption of water, alcohols and nitriles at the bridging Brønsted sites of H-ZSM-5, using both cluster and periodic models to account for the zeolite environment. Stabilization of the adsorption complexes results from hydrogen bonding between the guest molecule and the framework, as well as from embedding, i.e. van der Waals interactions with the pore walls. Large-cluster calculations with different DFT-methods, in particular B3LYP(-D), PBE(-D) M062X(-D) and ωB97X-D, are tested for their ability to reproduce the experimental heats of adsorption available in literature. (J. Phys. Chem. B 1997, 101, 3811-3817) A proper account of dispersion interactions is found to be crucial to describe the experimental trend across a series of adsorbates of increasing size, i.e. an increase in adsorption enthalpy by 10-15 kJ/mol for each additional carbon atom. The extended-cluster model is shown to offer an attractive alternative to periodic simulations on the entire H-ZSM-5 unit cell, resulting in virtually identical results for the final adsorption enthalpies. Comparing calculated stretch frequencies of the zeolite acid sites and the adsorbate functional groups with experimental IR-data additionally confirms the cluster approach provides an appropriate representation of the adsorption complexes.
 Open Access version available at UGent repository
Open Access version available at UGent repository