In-Depth Thermodynamic and Kinetic Analysis of Ethane Diffusion in ZIF-8
Abstract
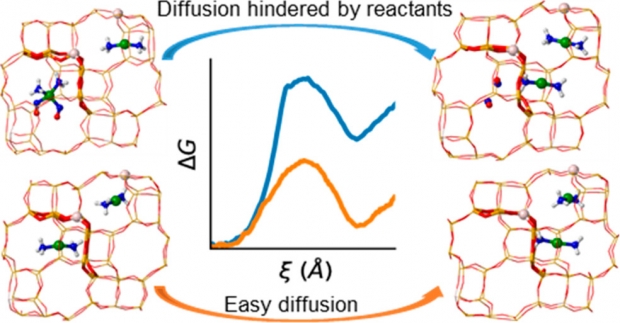
Flexible microporous ZIF-8 crystals show excellent separation behavior of small molecules such as ethaneand ethene. As such, hydrocarbon diffusion plays an essential role in the performance of these materials, yet determining accurate diffusion constants is nontrivial. Both ab initio and force-field based molecular dynamics simulations, coupled with umbrella sampling are applied in this work to characterize the diffusion of ethane in ZIF-8. Diffusion constants are extracted from the simulations by a combination of transition state theory and a random-walk hopping model, and are compared against experimentally measured values from literature. Ethane diffusion is a hindered process characterized by a transition state corresponding to an ethane molecule crossing the gate in between two neighboring cages formed by methylimidazole linkers. Free energy profiles of the diffusion process are derived and analyzed revealing the entropic nature of the barrier due to a counteracting of covalent host deformation energy and nonbonding host–guest interaction. A temperature analysis further confirms the entropic nature of the barrier and reveals an increased gate opening at increasing temperature. Finally, the loading dependency of diffusion is investigated revealing that increasing the ethane loading of the cages slightly slows down diffusion as a result of beneficial guest–guest interactions in the cages. Our findings yield essential elementary insight into how different molecular interactions influence the diffusion path of hydrocarbons throughout ZIF-8 crystals.
 Open Access version available at
Open Access version available at