Understanding Brønsted-Acid Catalyzed Monomolecular Reactions of Alkanes in Zeolite Pores by Combining Insights from Experiment and Theory
Abstract
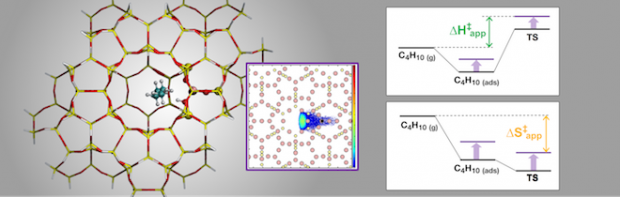
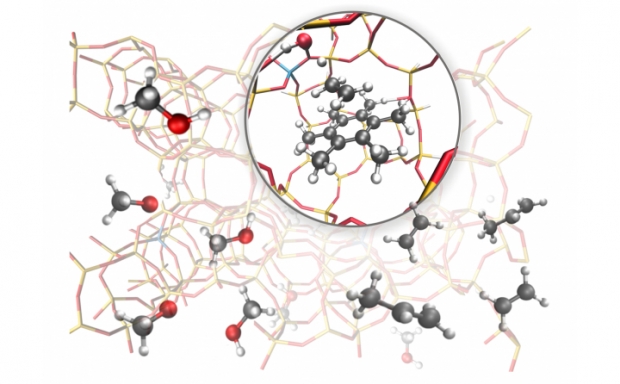
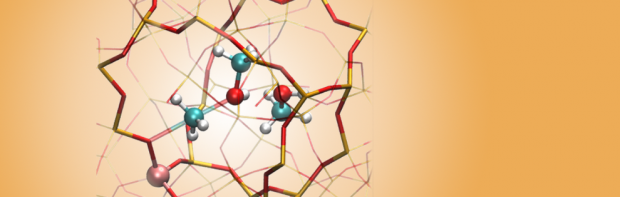
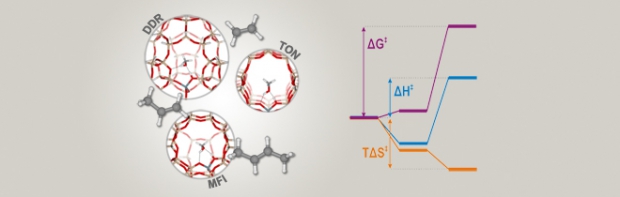
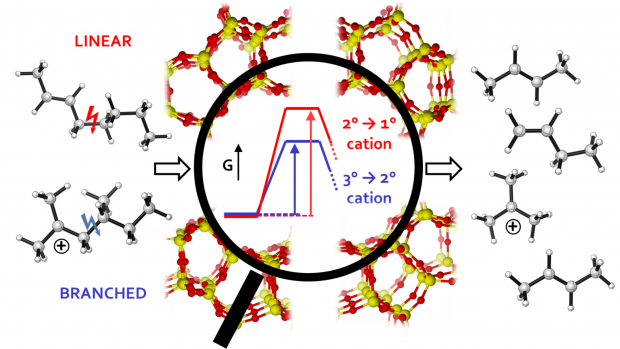
Acidic zeolites are effective catalysts for the cracking of large hydrocarbon molecules into lower molecular weight products required for transportation fuels. However, the ways in which the zeolite structure affects the catalytic activity at Brønsted protons are not fully understood. One way to characterize the influence of the zeolite structure on the catalysis is to study alkane cracking and dehydrogenation at very low conversion, conditions for which the kinetics are well defined. To understand the effects of zeolite structure on the measured rate coefficient (kapp), it is necessary to identify the equilibrium constant for adsorption into the reactant state (Kads-H+) and the intrinsic rate coefficient of the reaction (kint) at reaction temperatures, since kapp is proportional to the products of Kads-H+ and kint. We show that Kads-H+ cannot be calculated from experimental adsorption data collected near ambient temperature, but can, however, be estimated accurately from configurational-bias Monte Carlo (CBMC) simulations. Using monomolecular cracking and dehydrogenation of C3-C6 alkanes as an example, we review recent efforts aimed at elucidating the influence of the acid site location and the zeolite framework structure on the observed values of kapp and its components, Kads-H+ and kint.
 Open Access version available at
Open Access version available at