QuickFF: toward a generally applicable methodology to quickly derive accurate force fields for Metal-Organic Frameworks from ab initio input
Journal of Computational Chemistry
2015
A1
Published while none of the authors were employed at the CMM
The mechanical energy absorption ability of the highly flexible; MIL-53(Al) MOF material was explored using a combination of; experiments and molecular simulations. A pressure-induced transition; between the large pore and the closed pore forms of this solid; was revealed to be irreversible and associated with a relatively large; energy absorption capacity. Both features make MIL-53(Al) the first; potential MOF candidate for further use as a shock absorber.
 Open Access version available at UGent repository
Open Access version available at UGent repository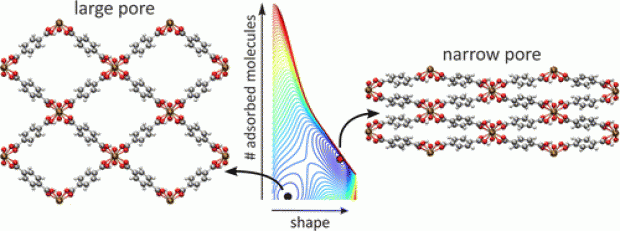
When adsorbing guest molecules, the porous metal-organic framework MIL-53(Cr) may vary its cell parameters drastically while retaining its crystallinity. A first approach to the thermodynamic analysis of this 'framework breathing' consists of comparing the osmotic potential in two distinct shapes only (large-pore and narrow-pore). In this paper, we propose a generic parametrized free energy model including three contributions: host free energy, guest-guest interactions, and host-guest interaction. Free energy landscapes may now be constructed scanning all shapes and any adsorbed amount of guest molecules. This allows to determine which shapes are the most stable states for arbitrary combinations of experimental control parameters, such as the adsorbing gas chemical potential, the external pressure, and the temperature. The new model correctly reproduces the structural transitions along the CO2 and CH4 isotherms. Moreover, our model successfully explains the adsorption versus desorption hysteresis as a consequence of the creation, stabilization, destabilization, and disappearance of a second free energy minimum under the assumptions of a first order phase transition and collective behavior. Our general thermodynamic description allows to decouple the gas chemical potential μ and mechanical pressure P as two independent thermodynamic variables and predict the complete (μ,P) phase diagram for CO2 adsorption in MIL-53(Cr). The free energy model proposed here is an important step towards a general thermodynamics description of flexible metal-organic frameworks.
An ecient protocol is presented to compensate for the basis set superposition error (BSSE) in DFT molecular dynamics (MD) simulations using localized Gaussian basis sets. We propose a classical correction term that can be added a posteriori to account for BSSE. It is tested to what extension this term will improve radial distribution functions (RDFs). The proposed term is pairwise between certain atoms in dierent molecules and was calibrated by tting reference BSSE data points computed with the counterpoise method. It is veried that the proposed exponential decaying functional form of the model is valid. This work focuses on hydrogen-bonded liquids, i.e. methanol, and more specic on the intermolecular hydrogen bond, but in principle the method is generally applicable on any type of interaction where BSSE is significant. We evaluated the relative importance of the Grimme-dispersion versus BSSE and found that they are of the same order of magnitude, but with an opposite sign. Upon introduction of the correction, the relevant RDFs, obtained from MD, have amplitudes equal to experiment.
Open Access version available at UGent repository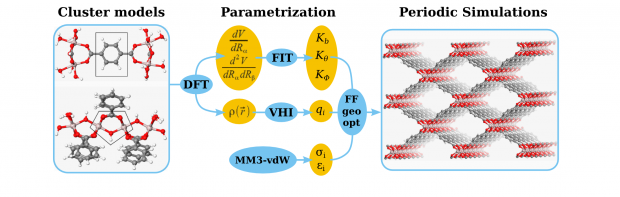
A force field is proposed for the flexible metal-organic framework MIL-53(Al), which is calibrated using density functional theory calculations on non-periodic clusters. The force field has three main contributions: an electrostatic term based on atomic charges derived with a modified Hirshfeld-I method, a van der Waals (vdW) term with parameters taken from the MM3 model and a valence force field whose parameters were estimated with a new methodology that uses the gradients and Hessian matrix elements retrieved from non-periodic cluster calculations. The new force field, predicts geometries and cell parameters that compare well with the experimental values both for the large and narrow pore phases. The energy profile along the breathing mode of the empty material reveals the existence of two minima, which confirms the intrinsic bistable behaviour of the MIL-53. Even without the stimulus of external guest molecules the material may transform from the large pore (lp) to the narrow pore (np) phase [Liu et al. JACS 2008, 120, 11813]. The relative stability of the two phases critically depends on the vdW parameters and MM3 dispersion interaction has the tendency to overstabilize the np phase.