Exploring the flexibility of MIL-47(V)-type materials using force field molecular dynamics simulations
Abstract
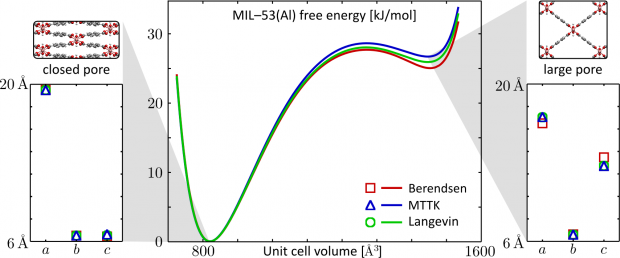
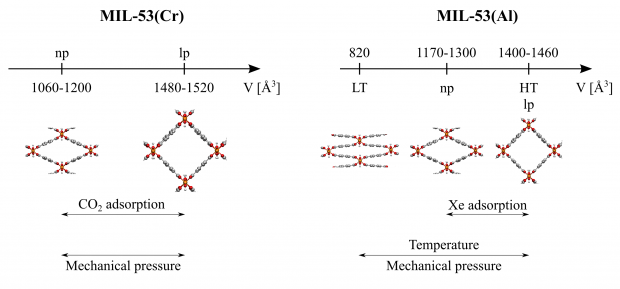
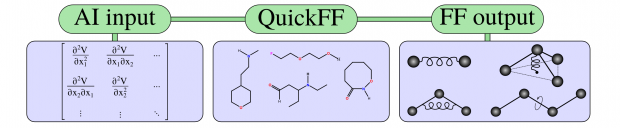
The flexibility of three MIL-47(V)-type materials (MIL-47, COMOC-2 and COMOC-3) has been explored by constructing the pressure-versus-volume and free energy-versus-volume curves at various temperatures ranging from 100 K to 400 K. This is done with first-principles based force fields using the recently proposed QuickFF parameterization protocol. Specific terms were added for the materials at hand to describe the asymmetry of the one-dimensional vanadium-oxide chain and to account for the flexibility of the organic linkers. The force fields are used in a series of molecular dynamics simulations at fixed volumes, but varying unit cell shapes. The three materials show a distinct pressure-versus-volume behavior, which underlines the ability to tune the mechanical properties by varying the linkers towards different applications such as nanosprings, dampers and shock absorbers.
 Open Access version available at UGent repository
Open Access version available at UGent repository