L. Vanduyfhuys
Cation−π Interactions Accelerate the Living Cationic Ring-Opening Polymerization of Unsaturated 2-Alkyl-2-oxazolines
Macromolecules
53, 10, 3832-3846
2020
A1
Abstract
Cation–dipole interactions were previously shown to have a rate-enhancing effect on the cationic ring-opening polymerization (CROP) of 2-oxazolines bearing a side-chain ester functionality. In line with this, a similar rate enhancement—via intermolecular cation−π interactions—was anticipated to occur when π-bonds are introduced into the 2-oxazoline side-chains. Moreover, the incorporation of π-bonds allows for facile postfunctionalization of the resulting poly(2-oxazoline)s with double and triple bonds in the side-chains via various click reactions. Herein, a combined molecular modeling and experimental approach was used to study the CROP reaction rates of 2-oxazolines with side-chains having varying degrees of unsaturation and side-chain length. The presence of cation−π interactions and the influence of the degree of unsaturation were initially confirmed by means of regular molecular dynamics simulations on pentameric systems. Furthermore, a combination of enhanced molecular dynamics simulations, static calculations, and a thorough analysis of the noncovalent interactions was performed to unravel to what extent cation−π interactions alter the reaction kinetics. Additionally, the observed trends were confirmed also in the presence of acetonitrile as solvent, in which experimentally the polymerization is performed. Most intriguingly, we found only a limited effect on the intrinsic reaction kinetics of the CROP and a preorganization effect in the reactive complex region. The latter effect was established by the unsaturated side-chains and the cationic center through a complex interplay between cation−π, π–π, π–induced dipole, and cation–dipole interactions. These findings led us to propose a two-step mechanism comprised of an equilibration step and a CROP reaction step. The influence of the degree of unsaturation, through a preorganization effect, on the equilibration step was determined with the following trend for the polymerization rates: n-ButylOx < ButenOx < ButynOx ≥ PentynOx. The trend was experimentally confirmed by determining the polymerization rate constants.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGold Open Access
Ab initio enhanced sampling kinetic study on MTO ethene methylation reaction
Journal of Catalysis
388, 38-51
2020
A1
Abstract
The methylation reaction of ethene with methanol over the Brønsted acidic ZSM-5 catalyst is one of theprototype reactions within zeolite catalysis for which experimental kinetic data is available. It is one ofthe premier reactions within the methanol-to-olefins process and has been the subject of extensive the-oretical testing to predict the reaction rates. Herein, we apply, for the first time, first principle moleculardynamics methods to determine the intrinsic reaction kinetics taking into account the full configurationalentropy. As chemical reactions are rare events, enhanced sampling methods are necessary to obtain suf-ficient sampling of the configurational space at the activated region. A plethora of methods is availablewhich depend on specific choices like the selection of collective variables along which the dynamics isenhanced. Herein, a thorough first principle molecular dynamics study is presented to determine thereaction kinetics via various enhanced MD techniques on an exemplary reaction within zeolite catalysisfor which reference theoretical and experimental data are available.
Green Open Access
Unraveling the thermodynamic conditions for negative gas adsorption in soft porous crystals
Communications Physics
2, 102
2019
A1
Abstract
Soft porous crystals (SPCs) are widely known for their intriguing properties and various counterintuitive phenomena such as negative linear compression, negative thermal expansion and negative gas adsorption (NGA). An intriguing case is the adsorption of methane in DUT-49 for which experimentally a drop in the amount of adsorbed particles was observed under increasing vapor pressure. It is yet unknown which specific systems can exhibit NGA under which thermodynamic conditions. Herein, a semi-analytical thermodynamic model is applied to determine the conditions required for NGA, including their sensitivity towards various system-specific parameters, and investigate the correlation with pressure-induced breathing. As such, it is found that certain non-breathing materials may exhibit breathing with NGA under application of a fixed mechanical pressure. Such meticulous control of multiple triggers for NGA can open the way to new applications such as tunable gas detection and pressure amplification.
Gold Open Access
Thermodynamic modeling of the selective adsorption of carbon dioxide over methane in the mechanically constrained breathing MIL-53(Cr)
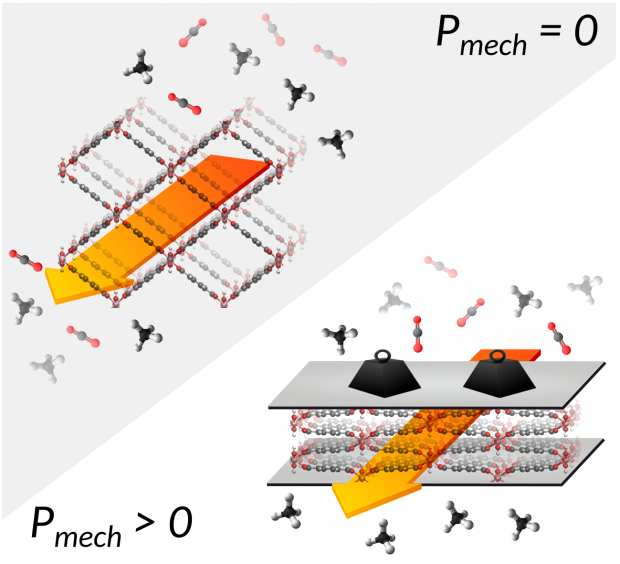
Advanced Theory and Simulations
1900124
2019
A1
Abstract
The coadsorption of CO2/CH4 in the breathing MIL-53(Cr) under the application of an additional mechanical pressure is investigated through the use of an extended thermodynamic mean-field model. The focus is on the breathing behavior, negative gas adsorption (NGA), and selective adsorption of CO2 as well as to what degree the application of mechanical pressure influences this behavior. To this end, phase diagrams, coadsorption isotherms are constructed and the CO 2 /CH 4 selectivity is computed in terms of the vapor pressure of methane and carbon dioxide as well as the mechanical pressure. As a result, it was found that NGA can be induced by coadsorption of CO2 /CH4 gas mixtures with certain molar compositions. Finally, a specific adsorption/desorption cycle, which includes the application of an additional mechanical pressure, is proposed to allow for an increased CO2/CH4 selectivity as well as for an expected less energy demanding CO2 desorption step.
Thermal Engineering of Metal-Organic Frameworks for Adsorption Applications: A Molecular Simulations Perspective
ACS Applied Materials & Interfaces
11 (42), 38697-38707
2019
A1
Abstract
Thermal engineering of metal-organic frameworks (MOFs) for adsorption-based applications is very topical in view of their industrial potential, especially since heat management and thermal stability have been identified as important obstacles. Hence, a fundamental understanding of the structural and chemical features underpinning their intrinsic thermal properties is highly sought-after. Herein, we investigate the nanoscale behavior of a diverse set of frameworks using molecular simulation techniques and critically compare properties such as thermal conductivity, heat capacity and thermal expansion with other material classes. Furthermore, we propose a hypothetical thermodynamic cycle to estimate the temperature rise associated with adsorption for the most important greenhouse and energy-related gases (CO2 and CH4). This macroscopic response on the heat of adsorption connects the intrinsic thermal properties with the adsorption properties, and allows us to evaluate their importance.
Pillared-layered metal-organic frameworks for mechanical energy storage applications
Journal of Materials Chemistry A
7 (39), 22663-22674
2019
A1
Abstract
Herein we explore the unique potential of pillared-layered metal–organic frameworks of the DMOF-1 family for mechanical energy storage applications. In this work, we theoretically predict for the guest-free DMOF-1 a new contracted phase by exerting an external mechanical pressure of more than 200 MPa with respect to the stable phase at atmospheric pressure. The breathing transition is accompanied by a very large volume contraction of about 40%. The high transition pressures and associated volume changes make these materials highly promising with an outstanding mechanical energy work. Furthermore, we show that changing the nature of the metal allows to tune the behavior under mechanical pressure. The various phases were revealed by a combination of periodic density-functional theory calculations, force field molecular dynamics simulations and mercury intrusion experiments for DMOF-1(Zn) and DMOF-1(Cu). The combined experimental and theoretical approach allowed to discover the potential of these materials for new technological developments.
Gold Open Access
Insight into the role of water on the methylation of hexamethylbenzene in H-SAPO-34 from first principle molecular dynamics simulations
ChemCatChem
11 (16), 3993-4010
2019
A1
Abstract
The methylation of hexamethylbenzene with methanol is one of the key reactions in the methanol‐to‐olefins hydrocarbon pool reaction cycle taking place over the industrially relevant H‐SAPO‐34 zeolite. This methylation reaction can occur either via a concerted or via a stepwise mechanism, the latter being the preferred pathway at higher temperatures. Herein, we systematically investigate how a complex reaction environment with additional water molecules and higher concentrations of Brønsted acid sites in the zeolite impacts the reaction mechanism. To this end, first principle molecular dynamics simulations are performed using enhanced sampling methods to characterize the reactants and products in the catalyst pores and to construct the free energy profiles. The most prominent effect of the dynamic sampling of the reaction path is the stabilization of the product region where water is formed, which can either move freely in the pores of the zeolite or be stabilized through hydrogen bonding with the other protic molecules. These protic molecules also stabilize the deprotonated Brønsted acid site, created due to the formation of the heptamethylbenzenium cation, via a Grotthuss‐type mechanism. Our results provide fundamental insight in the experimental parameters that impact the methylation of hexamethylbenzene in H‐SAPO‐34, especially highlighting and rationalizing the crucial role of water in one of the main reactions of the aromatics‐based reaction cycle.
Gold Open Access
The impact of lattice vibrations on the macroscopic breathing behavior of MIL-53(Al)
Zeitschrift für Kristallographie - Crystalline Materials
234 (7-8), 529-545
2019
A1
Abstract
The mechanism inducing the breathing in flexible metal-organic frameworks, such as MIL-53(Al), is still not fully understood. Herein, the influence of lattice vibrations on the breathing transition in MIL-53(Al) is investigated to gain insight in this phenomenon. Through solid-state density-functional theory calculations, the volume-dependent IR spectrum is computed together with the volume-frequency relations of all vibrational modes. Furthermore, important thermodynamic properties such as the Helmholtz free energy, the specific heat capacity, the bulk modulus, and the volumetric thermal expansion coefficient are derived via these volume-frequency relations using the quasi-harmonic approximation. The simulations expose a general volume-dependency of the vibrations with wavenumbers above 300 cm−1 due to their localized nature. In contrast, a diverse set of volume-frequency relations are observed for vibrations in the terahertz region (< 300 cm−1) containing the vibrations exhibiting collective behavior. Some terahertz vibrations display large frequency differences over the computed volume range, induced by either repulsion or strain effects, potentially triggering the phase transformation. Finally, the impact of the lattice vibrations on the thermodynamic properties is investigated. This reveals that the closed pore to large pore phase transformation in MIL-53(Al) is mainly facilitated by terahertz vibrations inducing rotations of the organic linker, while the large pore to closed pore phase transformation relies on two framework-specific soft modes.
Gold Open Access
Modeling Gas Adsorption in Flexible Metal–Organic Frameworks via Hybrid Monte Carlo / Molecular Dynamics Schemes
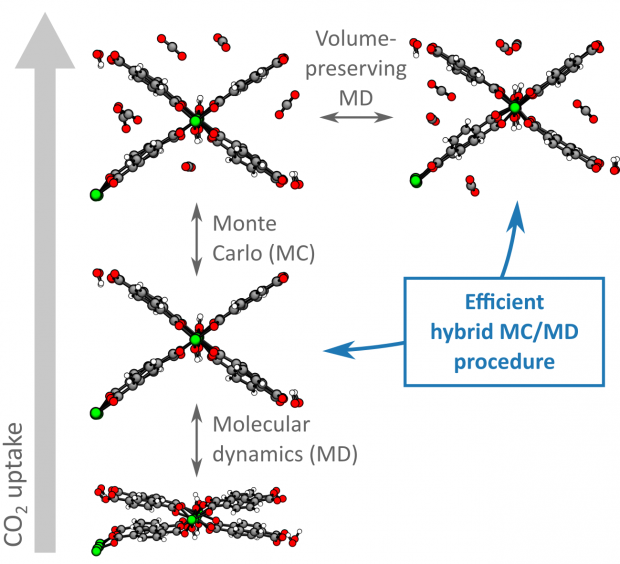
Advanced Theory and Simulations
2 (4), 1800177
2019
A1
Abstract
Herein, a hybrid Monte Carlo (MC)/molecular dynamics (MD) simulation protocol that properly accounts for the extraordinary structural flexibility of metal–organic frameworks (MOFs) is developed and validated. This is vital to accurately predict gas adsorption isotherms and guest‐induced flexibility of these materials. First, the performance of three recent models to predict adsorption isotherms and flexibility in MOFs is critically investigated. While these methods succeed in providing qualitative insight in the gas adsorption process in MOFs, their accuracy remains limited as the intrinsic flexibility of these materials is very hard to account for. To overcome this challenge, a hybrid MC/MD simulation protocol that is specifically designed to handle the flexibility of the adsorbent, including the shape flexibility, is introduced, thereby unifying the strengths of the previous models. It is demonstrated that the application of this new protocol to the adsorption of neon, argon, xenon, methane, and carbon dioxide in MIL‐53(Al), a prototypical flexible MOF, substantially decreases the inaccuracy of the obtained adsorption isotherms and predicted guest‐induced flexibility. As a result, this method is ideally suited to rationalize the adsorption performance of flexible nanoporous materials at the molecular level, paving the way for the conscious design of MOFs as industrial adsorbents.
Gold Open Access