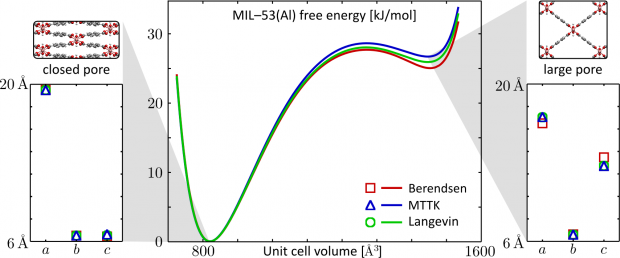
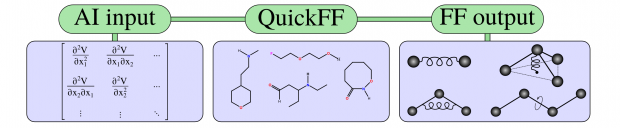
The ReaxFF reactive force-field: development, applications and future directions
Abstract
The reactive force-field (ReaxFF) interatomic potential is a powerful computational tool for exploring, developing and optimizing material properties. Methods based on the principles of quantum mechanics (QM), while offering valuable theoretical guidance at the electronic level, are often too computationally intense for simulations that consider the full dynamic evolution of a system. Alternatively, empirical interatomic potentials that are based on classical principles require significantly fewer computational resources, which enables simulations to better describe dynamic processes over longer timeframes and on larger scales. Such methods, however, typically require a predefined connectivity between atoms, precluding simulations that involve reactive events. The ReaxFF method was developed to help bridge this gap. Approaching the gap from the classical side, ReaxFF casts the empirical interatomic potential within a bond-order formalism, thus implicitly describing chemical bonding without expensive QM calculations. This article provides an overview of the development, application, and future directions of the ReaxFF method.
 Open Access version available at UGent repository
Open Access version available at UGent repository