Computation of charge distribution and electrostatic potential in silicates with the use of chemical potential equalization models
Abstract
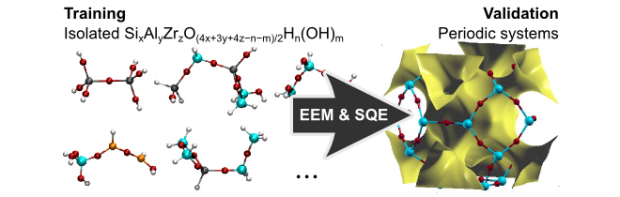
New parameters for the electronegativity equalization model (EEM) and the split-charge equilibration (SQE) model are calibrated for silicate materials, based on an extensive training set of representative isolated systems. In total, four calibrations are carried out, two for each model, either using iterative Hirshfeld (HI) charges or ESP grid data computed with Density Functional Theory (DFT) as a reference. Both the static (ground state) reference quantities and their responses to uniform electric fields are included in the fitting procedure. The EEM model fails to describe the response data, while the SQE model quantitatively reproduces all the training data. For the ESP-based parameters, we found that the reference ESP data are only useful at those grid points where the electron density is lower than 10-3 a.u. The density value correlates with a distance criterion used for selecting grid points in common ESP fitting schemes. All parameters are validated with DFT computations on an independent set of isolated systems (similar to the training set), and on a set of periodic systems including dense and microporous crystalline silica structures, zirconia, and zirconium silicate. Although the transferability of the parameters to new isolated systems poses no difficulties, the atomic hardness parameters in the HI-based models must be corrected to obtain accurate results for periodic systems. The SQE/ESP model permits the calculation of the ESP with similar accuracy in both isolated and periodic systems.
 Open Access version available at UGent repository
Open Access version available at UGent repository