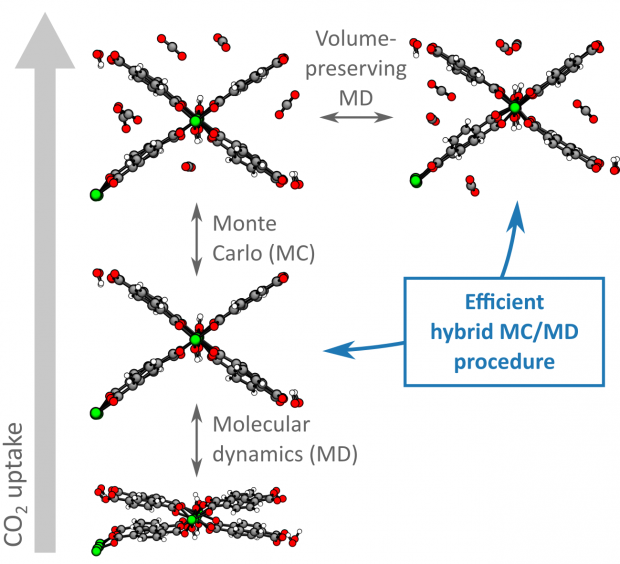
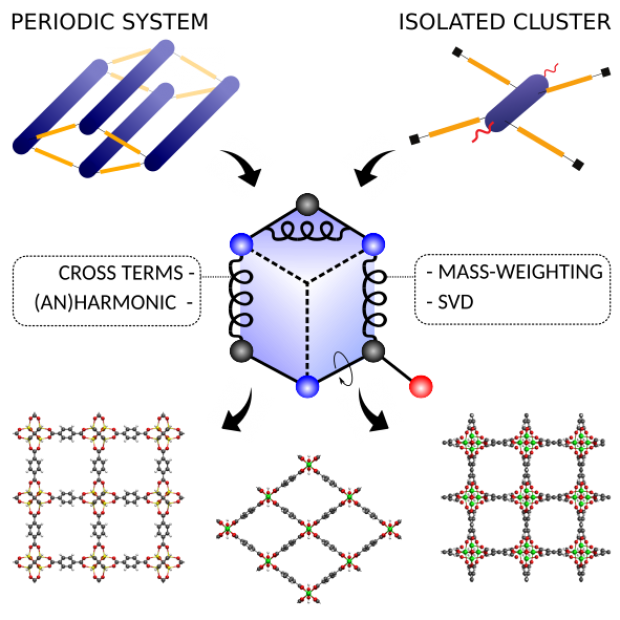
Charting the Complete Thermodynamic Landscape of Gas Adsorption for a Responsive Metal-Organic Framework
Abstract
New nanoporous materials have the ability to revolutionize adsorption and separation processes. In particular, materials with adaptive cavities have high selectivity and may display previously undiscovered phenomena, such as negative gas adsorption (NGA), in which gas is released from the framework upon an increase in pressure. Although the thermodynamic driving force behind this and many other counterintuitive adsorption phenomena have been thoroughly investigated in recent years, several experimental observations remain difficult to explain. This necessitates a comprehensive analysis of gas adsorption akin to the conformational free energy landscapes used to understand the function of proteins. We have constructed the complete thermodynamic landscape of methane adsorption on DUT-49. Traversing this complex landscape reproduces the experimentally observed structural transitions, temperature dependence, and the hysteresis between adsorption and desorption. The complete thermodynamic description presented here provides unparalleled insight into adsorption and provides a framework to understand other adsorbents that challenge our preconceptions.
 Open Access version available at
Open Access version available at