Abstract
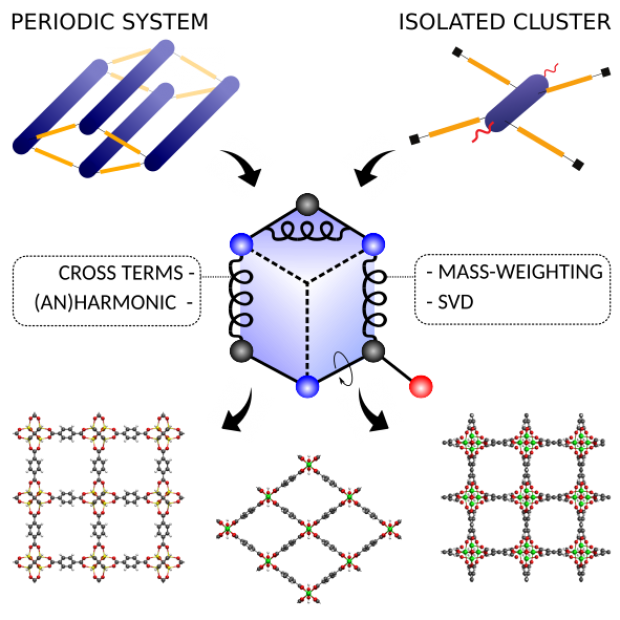
QuickFF was originally launched in 2015 to derive accurate force fields for isolated and complex molecular systems in a quick and easy way. Apart from the general applicability, the functionality was especially tested for metal-organic frameworks (MOFs), a class of hybrid materials consisting of organic and inorganic building blocks. Herein, we launch a new release of the QuickFF protocol which includes new major features to predict structural, vibrational, mechanical and thermal properties with greater accuracy, without compromising its robustness and transparant workflow. First, the ab initio data necessary for the fitting procedure may now also be derived from periodic models for the molecular system, as opposed to the earlier cluster-based models. This is essential for an accurate description of MOFs with one dimensional metal-oxide chains. Second, cross terms that couple internal coordinates (ICs) and anharmonic contributions for bond and bend terms are implemented. These features are essential for a proper description of vibrational and thermal properties. Third, the fitting scheme was modified to improve robustness and accuracy. The new features are tested on MIL-53(Al), MOF-5, CAU-13 and NOTT-300. As expected, periodic input data is proven to be essential for a correct description of structural, vibrational and thermodynamic properties of MIL-53(Al). Bulk moduli and thermal expansion coefficients of MOF-5 are very accurately reproduced by static and dynamic simulations using the newly derived force fields which include cross terms and anharmonic corrections. For the flexible materials CAU-13 and NOTT-300, the transition pressure is accurately
predicted provided cross terms are taken into account.
 Open Access version available at UGent repository
Open Access version available at UGent repository