D.E.P. Vanpoucke, J. Jaeken, S. De Baerdemacker, K. Lejaeghere, V. Van Speybroeck
Beilstein Journal of Nanotechnology
Abstract
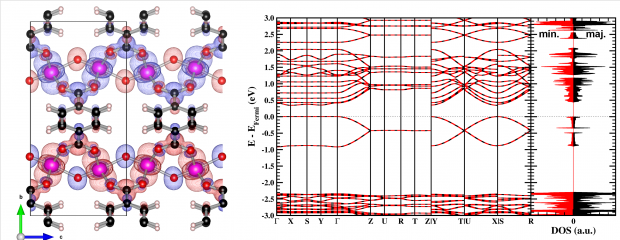
The geometric and electronic structure of the MIL-47(V) metal-organic framework (MOF) is investigated by using ab initio density functional theory (DFT) calculations. Special focus is placed on the relation between the spin configuration and the properties of the MOF. The ground state is found to be antiferromagnetic, with an equilibrium volume of 1554.70 Å3. The transition pressure of the pressure-induced large-pore-to-narrow-pore phase transition is calculated to be 82 MPa and 124 MPa for systems with ferromagnetic and antiferromagnetic chains, respectively. For a mixed system, the transition pressure is found to be a weighted average of the ferromagnetic and antiferromagnetic transition pressures. Mapping DFT energies onto a simple-spin Hamiltonian shows both the intra- and inter-chain coupling to be antiferromagnetic, with the latter coupling constant being two orders of magnitude smaller than the former, suggesting the MIL-47(V) to present quasi-1D behavior. The electronic structure of the different spin configurations is investigated and it shows that the band gap position varies strongly with the spin configuration. The valence and conduction bands show a clear V d-character. In addition, these bands are flat in directions orthogonal to VO6 chains, while showing dispersion along the the direction of the VO6 chains, similar as for other quasi-1D materials
 Open Access version available at
Open Access version available at