K. Lejaeghere

Towards accurate processing-structure-property links using deep learning
The ABINIT project: Impact, environment and recent developments
Computer Physics Communications
248, 107042
2020
A1
Abstract
Abinit is a material- and nanostructure-oriented package that implements density-functional theory (DFT) and many-body perturbation theory (MBPT) to find, from first principles, numerous properties including total energy, electronic structure, vibrational and thermodynamic properties, different dielectric and non-linear optical properties, and related spectra. In the special issue to celebrate the 40th anniversary of CPC, published in 2009, a detailed account of Abinit was included [Gonze et al. (2009)], and has been amply cited. The present article comes as a follow-up to this 2009 publication. It includes an analysis of the impact that Abinit has had, through for example the bibliometric indicators of the 2009 publication. Links with several other computational materials science projects are described. This article also covers the new capabilities of Abinit that have been implemented during the last three years, complementing a recent update of the 2009 article published in 2016. Physical and technical developments inside the abinit application are covered, as well as developments provided with the Abinit package, such as the multibinit and a-tdep projects, and related Abinit organization developments such as AbiPy . The new developments are described with relevant references, input variables, tests, and tutorials.
Optical Properties of Isolated and Covalent Organic Framework-Embedded Ruthenium Complexes
Journal of Physical Chemistry A
123 (32), 6854-6867
2019
A1
Abstract
Heterogenization of RuL3 complexes on a support with proper anchor points provides a route toward design of green catalysts. In this paper, Ru(II) polypyridyl complexes are investigated with the aim to unravel the influence on the photocatalytic properties of varying nitrogen content in the ligands and of embedding the complex in a triazine-based covalent organic framework. To provide fundamental insight into the electronic mechanisms underlying this behavior, a computational study is performed. Both the ground and excited state properties of isolated and anchored ruthenium complexes are theoretically investigated by means of density functional theory and time-dependent density functional theory. Varying the ligands among 2,2′-bipyridine, 2,2′-bipyrimidine, and 2,2′-bipyrazine allows us to tune to a certain extent the optical gaps and the metal to ligand charge transfer excitations. Heterogenization of the complex within a CTF support has a significant effect on the nature and energy of the electronic transitions. The allowed transitions are significantly red-shifted toward the near IR region and involve transitions from states localized on the CTF toward ligands attached to the ruthenium. The study shows how variations in ligands and anchoring on proper supports allows us to increase the range of wavelengths that may be exploited for photocatalysis.
Gold Open Access
Thermal unequilibrium of strained black CsPbI3 thin films
Science
365 (6454), 679-684
2019
A1
Abstract
The high-temperature all-inorganic CsPbI3 perovskite black phase is metastable relative to its yellow non-perovskite phase, at room temperature. Since only the black phase is optically active, this represents an impediment for the use of CsPbI3 in optoelectronic devices. We report the use of substrate clamping and biaxial strain to render stable, at room temperature, black phase CsPbI3 thin films. We used synchrotron-based grazing incidence wide angle x-ray scattering to track the introduction of crystal distortions and strain-driven texture formation within black CsPbI3 thin films when they were cooled following annealing at 330°C. The thermal stability of black CsPbI3 thin films is vastly improved by the strained interface, a response verified by ab initio thermodynamic modelling.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGold Open Access
Electronic properties of heterogenized Ru(II) polypyridyl photoredox complexes on covalent triazine frameworks
Journal of Materials Chemistry A
7, 8433-8442
2019
A1
Abstract
Ru(II) polypyridyl complexes have been successful for a wide range of photoredox applications thanks to their efficient light-induced metal-to-ligand charge transfer. Using the computational framework of density-functional theory, we report how these complexes can be anchored onto covalent triazine frameworks while maintaining their favorable electronic properties. We moreover show that variation of the nitrogen content of the framework linkers or complex ligands endows the heterogenized catalyst with a unique versatility, spanning a wide range of absorption characteristics and redox potentials. By judiciously choosing the catalyst building blocks, it is even possible to selectively guide the charge transfer toward either the scaffold or the accessible pore sites. Rational design of sustainable and efficient photocatalysts thus comes within reach.
A first-principles reassessment of the Fe-N phase diagram in the low-nitrogen limit
Journal of Alloys and Compounds
775, 758-768
2019
A1
Exploring lanthanide doping in UiO-66: a combined experimental and computational study of the electronic structure
Tuning the balance between dispersion and entropy to design temperature-responsive flexible metal-organic frameworks
Missing linkers: an alternative pathway to UiO-66 electronic structure engineering
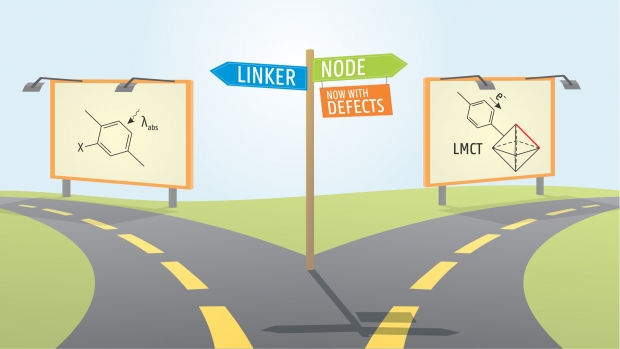
Chemistry of Materials
29 (7), 3006–3019
2017
A1
Abstract
UiO-66 is a promising metal-organic framework for photocatalytic applications. However, the ligand-to-metal charge transfer of an excited electron is inefficient in the pristine material. Herein we assess the influence of missing linker defects on the electronic structure of UiO-66 and discuss their ability to improve ligand-to-metal charge transfer. Using a new defect classification system, which is transparent and easily extendable, we identify the most promising photocatalysts by considering both relative stability and electronic structure. We find the properties of UiO-66 defect structures largely to depend on the coordination of the constituent nodes, and the nodes with the strongest local distortions to alter the electronic structure most. Defects hence provide an alternative pathway to tune UiO-66 for photocatalytic purposes, besides linker modification and node metal substitution. In addition, the decomposition of MOF properties into node- and linker-based behavior is more generally valid, so we propose orthogonal electronic structure tuning as a paradigm in MOF electronic structure engineering.
Open Access version available at UGent repositoryGold Open Access