Dynamic interplay between defective UiO-66 and protic solvents in activated processes
Abstract
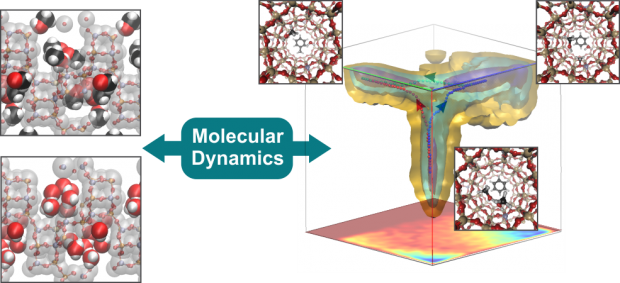
UiO‐66, composed by Zr‐oxide inorganic bricks [Zr6(μ3‐O)4(μ3‐OH)4] and organic terephthalate linkers, is one of the most studied metal–organic frameworks (MOFs) due to its exceptional thermal, chemical, and mechanical stability. Thanks to its high connectivity, the material can withstand structural deformations during activation processes such as linker exchange, dehydration, and defect formation. These processes do alter the zirconium coordination number in a dynamic way, creating open metal sites for catalysis and thus are able to tune the catalytic properties. In this work, it is shown, by means of first‐principle molecular‐dynamics simulations at operating conditions, how protic solvents may facilitate such changes in the metal coordination. Solvent can induce structural rearrangements in the material that can lead to undercoordinated but also overcoordinated metal sites. This is demonstrated by simulating activation processes along well‐chosen collective variables. Such enhanced MD simulations are able to track the intrinsic dynamics of the framework at realistic conditions.
 Open Access version available at
Open Access version available at