High-rate nanofluidic energy absorption in porous zeolitic frameworks

Abstract
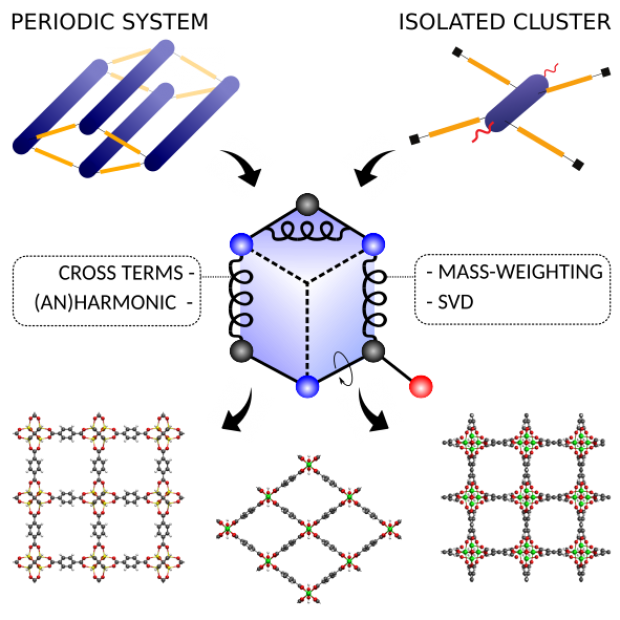
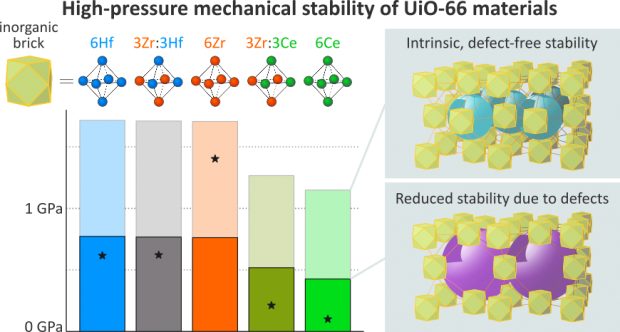
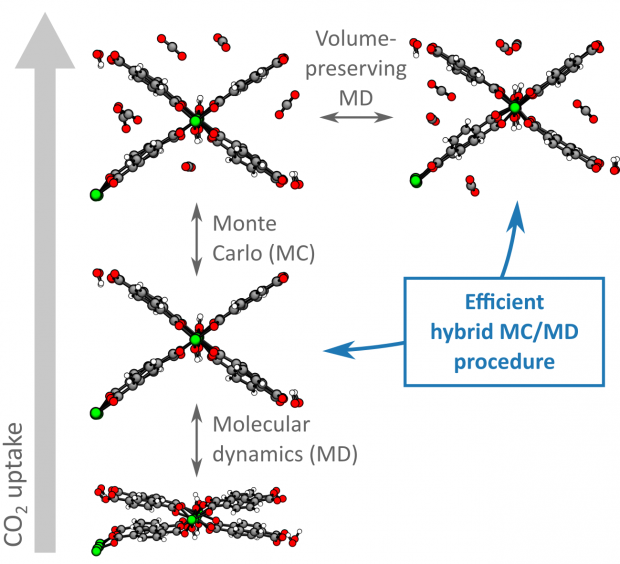
Optimal mechanical impact absorbers are reusable and exhibit high specific energy absorption. The forced intrusion of liquid water in hydrophobic nanoporous materials, such as zeolitic imidazolate frameworks (ZIFs), presents an attractive pathway to engineer such systems. However, to harness their full potential, it is crucial to understand the underlying water intrusion and extrusion mechanisms under realistic, high-rate deformation conditions. Here, we report a critical increase of the energy absorption capacity of confined water-ZIF systems at elevated strain rates. Starting from ZIF-8 as proof-of-concept, we demonstrate that this attractive rate dependence is generally applicable to cage-type ZIFs but disappears for channel-containing zeolites. Molecular simulations reveal that this phenomenon originates from the intrinsic nanosecond timescale needed for critical-sized water clusters to nucleate inside the nanocages, expediting water transport through the framework. Harnessing this fundamental understanding, design rules are formulated to construct effective, tailorable and reusable impact energy absorbers for challenging new applications.

 Open Access version available at
Open Access version available at