Complex reaction environments and competing reaction mechanisms in zeolite catalysis: insights from advanced molecular dynamics

Abstract
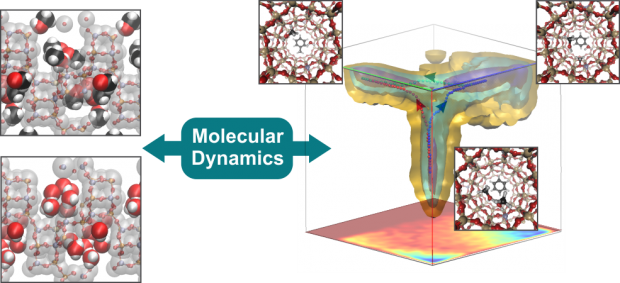
The methanol to olefins process is a show case example of complex zeolite-catalyzed chemistry. At real operating conditions, many factors such as framework flexibility, adsorption of various guest molecules and competitive reaction pathways, affect reactivity. In this paper we show the strength of first principle molecular dynamics techniques to capture this complexity by means of two case studies. Firstly, the adsorption behavior of methanol and water in H-SAPO-34 at 350 °C is investigated. Hereby we observed an important degree of framework flexibility and proton mobility. Secondly, we studied the methylation of benzene by methanol via a competitive direct and stepwise pathway in the AFI topology. Both case studies clearly show that a first principle molecular dynamics approach enables to obtain unprecedented insights into zeolite-catalyzed reactions at the nanometer scale.
 Open Access version available at UGent repository
Open Access version available at UGent repository