Bipyridine-Based Nanosized Metal–Organic Framework with Tunable Luminescence by a Postmodification with Eu(III): An Experimental and Theoretical Study
Abstract
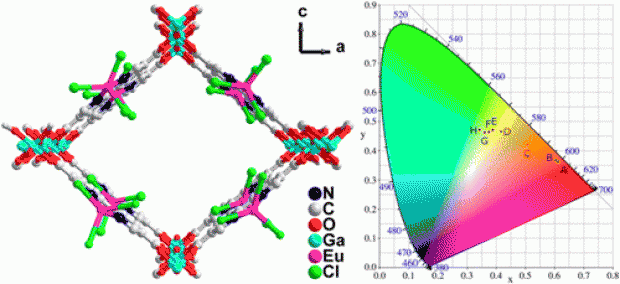
A gallium 2,2′-bipyridine-5,5′-dicarboxylate metal-organic framework, Ga(OH)(bpydc), denoted as COMOC-4 (COMOC = Center for Ordered Materials, Organometallics and Catalysis, Ghent University) has been synthesized via solvothermal synthesis procedure. The structure has the topology of an aluminum 2,2′-bipyridine-5,5′-dicarboxylate, the so-called MOF-253. TEM and SEM micrographs show the COMOC-4 crystals are formed in nanoplates with uniform size of 30-50 nm. The UV-Vis spectra of COMOC-4 in methanol solution show maximal electronic absorption at 307 nm. This results from linker to linker transitions as elucidated by time-dependent density functional theory simulations on the linker and COMOC-4 cluster models. When excited at 400 nm, COMOC-4 displays an emission band centered at 542 nm. Upon immersion in different solvents, the emission band for the framework is shifted in the range of 525~548 nm, depending on the solvent. After incorporating Eu3+ cations, the emission band of the framework is shifted to even shorter wavelengths (505 nm). By varying the excitation wavelengths from 250 to 400 nm, we can fine-tune the emission from red to yellowish green in the CIE diagram. The luminescence behavior of Eu3+ cations is well preserved and the solid state luminescence lifetimes of λ1 = 45 µs (35.4 %) and λ2 = 162 µs (64.6 %) are observed.

 Open Access version available at
Open Access version available at