Automated generation of radical species in crystalline carbohydrate using ab initio MD simulations
Abstract
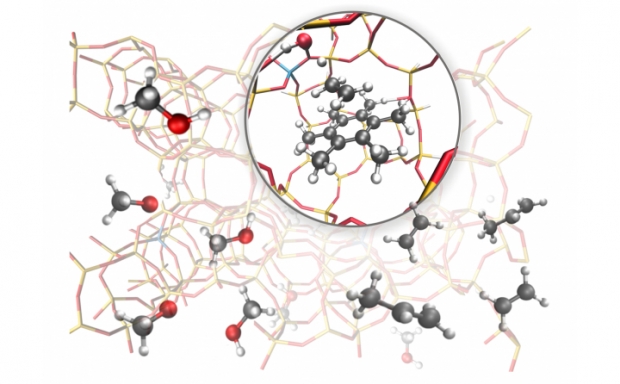
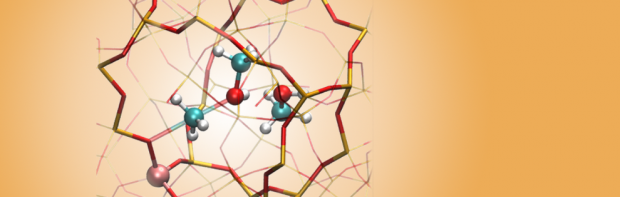
As the chemical structures of radiation damaged molecules may vary greatly from their undamaged counterparts, investigation and description of radiation damaged structures is commonly biased by the researcher. Radical formation from ionizing radiation in crystalline α-L-rhamnose monohydrate has been investigated using a new method where the selection of radical structures is unbiased by the researcher. The method is based on using ab initio molecular dynamics (MD) studies to investigate how ionization damage can form, change and move. Diversity in the radical production is gained by using different points on the potential energy surface of the intact crystal as starting points for the ionizations and letting the initial velocities of the nuclei after ionization be generated randomly. 160 ab initio MD runs produced 12 unique radical structures for investigation. Out of these, 7 of the potential products have never previously been discussed, and 3 products are found to match with radicals previously observed by electron magnetic resonance experiments
 Open Access version available at UGent repository
Open Access version available at UGent repository