Thermal unequilibrium of strained black CsPbI3 thin films
Abstract
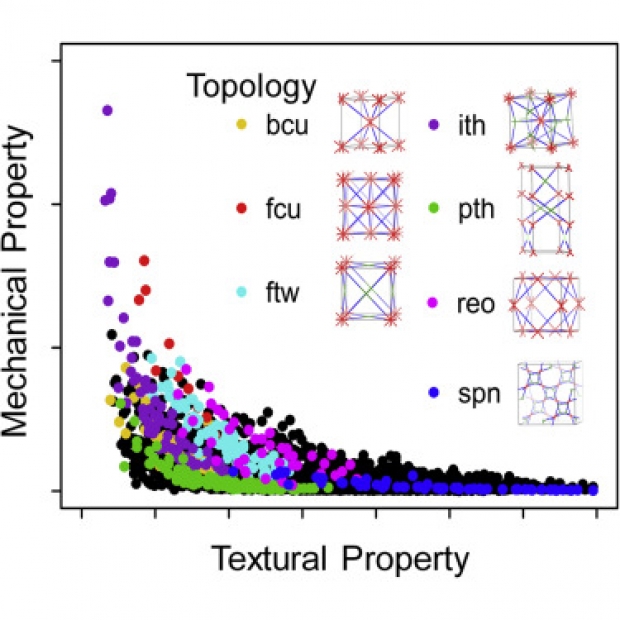
The high-temperature all-inorganic CsPbI3 perovskite black phase is metastable relative to its yellow non-perovskite phase, at room temperature. Since only the black phase is optically active, this represents an impediment for the use of CsPbI3 in optoelectronic devices. We report the use of substrate clamping and biaxial strain to render stable, at room temperature, black phase CsPbI3 thin films. We used synchrotron-based grazing incidence wide angle x-ray scattering to track the introduction of crystal distortions and strain-driven texture formation within black CsPbI3 thin films when they were cooled following annealing at 330°C. The thermal stability of black CsPbI3 thin films is vastly improved by the strained interface, a response verified by ab initio thermodynamic modelling.
 Open Access version available at UGent repository
Open Access version available at UGent repository
-zeolites is a mobile complex resembling a homogeneous catalyst, tethered by Coulomb interaction to the framework. The complexes are created in situ by coordination of ethene to Ni and lead to second order rate dependence on ethene pressure. Dimerization occurs by the Cossee-Arlman mechanism through transition states having the favoured square-planar coordination of Ni.")