Abstract
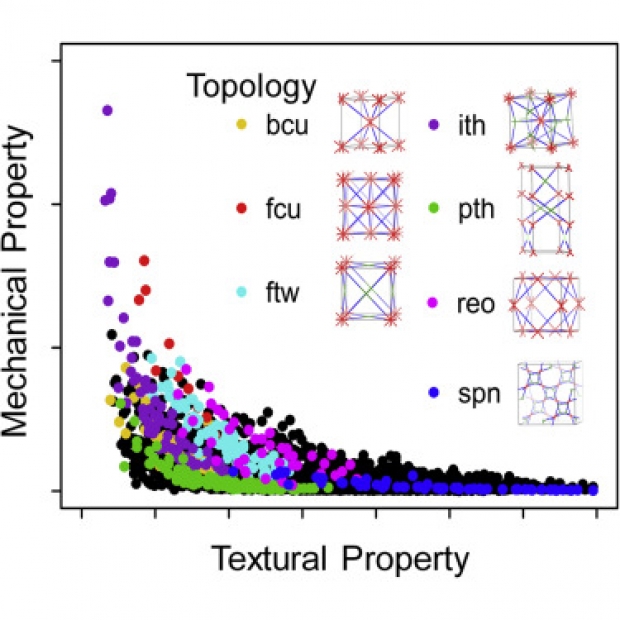
Assessing the mechanical stability of metal-organic frameworks (MOFs) is critical to bring these materials to any application. Here, we derive the first interactive map of the structure-mechanical landscape of MOFs by performing a multi-level computational analysis. First, we used high-throughput molecular simulations for 3,385 MOFs containing 41 distinct network topologies. Second, we developed a freely available machine-learning algorithm to automatically predict the mechanical properties of MOFs. For distinct regions of the high-throughput space, in-depth analysis based on in operando molecular dynamics simulations reveals the loss-of-crystallinity pressure within a given topology. The overarching mechanical screening approach presented here reveals the sensitivity on structural parameters such as topology, coordination characteristics and the nature of the building blocks, and paves the way for computational as well as experimental researchers to assess and design MOFs with enhanced mechanical stability to accelerate the translation of MOFs to industrial applications.