V. Van Speybroeck
N‐rich porous polymer with isolated Tb3+‐ions displays unique temperature dependent behavior through the absence of thermal quenching
Chemistry - A European Journal
26 (67), 15596-15604
2020
A1
Abstract
The challenge of measuring fast moving or small scale samples is based on the absence of contact betw een sample and sensor. Grafting lanthanides onto hybrid materials arises as one of the most promising accurate techniques to obtain noninvasive thermometers. In this w ork, a novel bipyridine based Porous Organic Polymer (bpyDATPOP) wasinvestigatedastemperaturesensorafter grafting w ith Eu(acac) 3 and Tb(acac) 3 complexes. The bpyDAT POP successfully showed temperature dependent behavior in the 10 ‐ 310 K range, proving the potential of amorphous, porous organic framew orks. More intriguingly, w e observed unique temperature dependent behavior; instead of the standard observed change in emission as a result of a change in temperature for both Eu 3+ and Tb 3+ , the emission spectrumof Tb 3+ remained constant. This w ork provides framework‐ and energy‐based explanations for the observed phenomenon. The conjugation in the bpyDAT POP framew ork is interrupted, creating energetically isolated Tb 3+ environments. Energy transferfromTb 3+ toEu 3+ isthereforeabsent,norenergybacktransfer from Tb 3+ to bpyDAT POP ligand (i.e. no thermal quenching) is detected.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGold Open Access
Cation−π Interactions Accelerate the Living Cationic Ring-Opening Polymerization of Unsaturated 2-Alkyl-2-oxazolines
Macromolecules
53, 10, 3832-3846
2020
A1
Abstract
Cation–dipole interactions were previously shown to have a rate-enhancing effect on the cationic ring-opening polymerization (CROP) of 2-oxazolines bearing a side-chain ester functionality. In line with this, a similar rate enhancement—via intermolecular cation−π interactions—was anticipated to occur when π-bonds are introduced into the 2-oxazoline side-chains. Moreover, the incorporation of π-bonds allows for facile postfunctionalization of the resulting poly(2-oxazoline)s with double and triple bonds in the side-chains via various click reactions. Herein, a combined molecular modeling and experimental approach was used to study the CROP reaction rates of 2-oxazolines with side-chains having varying degrees of unsaturation and side-chain length. The presence of cation−π interactions and the influence of the degree of unsaturation were initially confirmed by means of regular molecular dynamics simulations on pentameric systems. Furthermore, a combination of enhanced molecular dynamics simulations, static calculations, and a thorough analysis of the noncovalent interactions was performed to unravel to what extent cation−π interactions alter the reaction kinetics. Additionally, the observed trends were confirmed also in the presence of acetonitrile as solvent, in which experimentally the polymerization is performed. Most intriguingly, we found only a limited effect on the intrinsic reaction kinetics of the CROP and a preorganization effect in the reactive complex region. The latter effect was established by the unsaturated side-chains and the cationic center through a complex interplay between cation−π, π–π, π–induced dipole, and cation–dipole interactions. These findings led us to propose a two-step mechanism comprised of an equilibration step and a CROP reaction step. The influence of the degree of unsaturation, through a preorganization effect, on the equilibration step was determined with the following trend for the polymerization rates: n-ButylOx < ButenOx < ButynOx ≥ PentynOx. The trend was experimentally confirmed by determining the polymerization rate constants.
Open Access version available at UGent repositoryGold Open Access
Ab initio enhanced sampling kinetic study on MTO ethene methylation reaction
Journal of Catalysis
388, 38-51
2020
A1
Abstract
The methylation reaction of ethene with methanol over the Brønsted acidic ZSM-5 catalyst is one of theprototype reactions within zeolite catalysis for which experimental kinetic data is available. It is one ofthe premier reactions within the methanol-to-olefins process and has been the subject of extensive the-oretical testing to predict the reaction rates. Herein, we apply, for the first time, first principle moleculardynamics methods to determine the intrinsic reaction kinetics taking into account the full configurationalentropy. As chemical reactions are rare events, enhanced sampling methods are necessary to obtain suf-ficient sampling of the configurational space at the activated region. A plethora of methods is availablewhich depend on specific choices like the selection of collective variables along which the dynamics isenhanced. Herein, a thorough first principle molecular dynamics study is presented to determine thereaction kinetics via various enhanced MD techniques on an exemplary reaction within zeolite catalysisfor which reference theoretical and experimental data are available.
Green Open Access
Elucidating the promotional effect of a covalent triazine framework in aerobic oxidation
Applied Catalysis B: Environmental
269, 118769
2020
A1
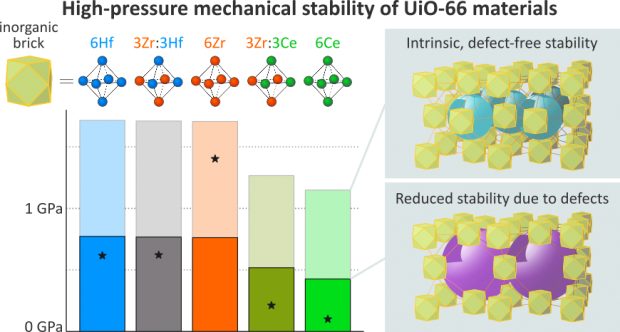
Charting the Metal-Dependent High-Pressure Stability of Bimetallic UiO-66 Materials
ACS Materials Letters
2 (4), 438-445
2020
A1
Abstract
In theory, bimetallic UiO-66(Zr:Ce) and UiO-66(Zr:Hf) metal-organic frameworks (MOFs) are extremely versatile and attractive nanoporous materials as they combine the high catalytic activity of UiO-66(Ce) or UiO-66(Hf) with the outstanding stability of UiO-66(Zr). Using in situ high-pressure powder X-ray diffraction, however, we observe that this expected mechanical stability is not achieved when incorporating cerium or hafnium in UiO-66(Zr). This observation is akin to the earlier observed reduced thermal stability of UiO-66(Zr:Ce) compounds. To elucidate the atomic origin of this phenomenon, we chart the loss-of-crystallinity pressures of 22 monometallic and bimetallic UiO-66 materials and systematically isolate their intrinsic mechanical stability from their defect-induced weakening. This complementary experimental/computational approach reveals that the intrinsic mechanical stability of these bimetallic MOFs decreases nonlinearly upon cerium incorporation but remains unaffected by the zirconium:hafnium ratio. Additionally, all experimental samples suffer from defect-induced weakening, a synthesis-controlled effect that is observed to be independent of their intrinsic stability.
Gold Open Access
Light Olefin Diffusion during the MTO Process on H-SAPO-34: a Complex Interplay of Molecular Factors

JACS (Journal of the American Chemical Society)
142 (13), 6007-6017
2020
A1
Abstract
The methanol-to-olefins process over H-SAPO-34 is characterized by its high shape selectivity toward light olefins. The catalyst is a supramolecular system consisting of nanometer-sized inorganic cages, decorated by Brønsted acid sites, in which organic compounds, mostly methylated benzene species, are trapped. These hydrocarbon pool species are essential to catalyze the methanol conversion but may also clog the pores. As such, diffusion of ethene and propene plays an essential role in determining the ultimate product selectivity. Enhanced sampling molecular dynamics simulations based on either force fields or density functional theory are used to determine how molecular factors influence the diffusion of light olefins through the 8-ring windows of H-SAPO-34. Our simulations show that diffusion through the 8-ring in general is a hindered process, corresponding to a hopping event of the diffusing molecule between neighboring cages. The loading of different methanol, alkene, and aromatic species in the cages may substantially slow down or facilitate the diffusion process. The presence of Brønsted acid sites in the 8-ring enhances the diffusion process due to the formation of a favorable π-complex host–guest interaction. Aromatic hydrocarbon pool species severely hinder the diffusion and their spatial distribution in the zeolite crystal may have a significant effect on the product selectivity. Herein, we unveil how molecular factors influence the diffusion of light olefins in a complex environment with confined hydrocarbon pool species, high olefin loadings, and the presence of acid sites by means of enhanced molecular dynamics simulations under operating conditions.
Atomistic insight in the flexibility and heat transport properties of the stimuli-responsive metal-organic framework MIL-53(Al) for water-adsorption applications using molecular simulations
Faraday Discussions
225, 301-323
2021
A1
Abstract
To exploit the full potential of metal-organic frameworks as solid adsorbents in water-adsorption applications, many challenges remain to be solved. A more fundamental insight into the properties of the host material and the influence water exerts on them can be obtained by performing molecular simulations. In this work, the prototypical flexible MIL-53(Al) framework is modelled using advanced molecular dynamics simulations. For different water loadings, the presence of water is shown to affect the relative stability of MIL-53(Al), triggering a phase transition from the narrow-pore to the large-pore phase at the highest considered loading. Furthermore, the effect of confinement on the structural organisation of the water molecules is also examined for different pore volumes of MIL-53(Al). For the framework itself, we focus on the thermal conductivity, as this property plays a decisive role in the efficiency of adsorption-based technologies, due to the energy-intensive adsorption and desorption cycles. To this end, the heat transfer characteristics of both phases of MIL-53(Al) are studied, demonstrating a strong directional dependence for the thermal conductivity.
Gold Open Access
Engineering a highly defective stable UiO-66 with tunable Lewis-Brønsted acidity - The role of the hemilabile linker
JACS (Journal of the American Chemical Society)
142 (6), 3174-3183
2020
A1
Abstract
The stability of metal-organic frameworks (MOFs) typically decreases with an increasing number of defects, limiting the number of defects that can be created and limiting catalytic and other applications. Herein, we use a hemilabile (Hl) linker to create up to maximum 6 defects per cluster in UiO-66. We have synthesized hemilabile UiO-66 (Hl-UiO-66) using benzene dicarboxylate (BDC) as linker and 4-sulfonatobenzoate (PSBA) as the hemilabile linker. The PSBA acts not only as a modulator to create defects, but also as a co-ligand that enhances the stability of the resulting defective framework. Furthermore, upon a post-synthetic treatment in H2SO4, the average number of defects increases to the optimum of six missing BDC linkers per cluster (3 per formula unit), leaving the Zr-nodes on average 6-fold coordinated. Remarkably, the thermal stability of the materials further increases upon this treatment. Periodic density functional theory calculations confirm that the hemilabile ligands strengthen this highly defective structure by several stabilizing interactions. Finally, the catalytic activity of the obtained materials is evaluated in the acid-catalyzed isomerization of α-pinene oxide. This reaction is particularly sensitive to the Brønsted or Lewis acid sites in the catalyst. In comparison to the pristine UiO-66, which mainly possesses Brønsted acid sites, the Hl-UiO-66 and the post-synthetically treated Hl-UiO-66 structures exhibited a higher Lewis acidity and an enhanced activity and selectivity. This is further explored by CD3CN spectroscopic sorption experiments. We have shown that by tuning the number of defects in UiO-66 using PSBA as the hemilabile linker, one can achieve highly defective and stable MOFs and easily control the Brønsted to Lewis acid ratio in the materials, and thus their catalytic activity and selectivity.
The potential of anthocyanins from blueberries as a natural dye for cotton: A combined experimental and theoretical study
Dyes and Pigments
176, 108180
2020
A1
Abstract
Natural dyes might be more environmentally sustainable compared to their synthetic counterparts, however in general their performance is worse. Therefore, typically metallic mordants are applied to improve the natural dye's affinity towards substrates, but this is not a suitable technique in a ‘green story’. In this paper, we test the potential of using anthocyanins from blueberry waste for dyeing cotton with biomordants, which are selected to tailor the intermolecular interactions such as hydrogen bonds, ionic bonds and π-π interactions with the dye molecule. In the experimental part, parameters during extraction and dyeing were optimized (e.g. temperature, pH, dyeing time and concentration). The effect of the (bio)mordants was monitored by Fourier transform infrared spectroscopy, spectrophotometric measurements and standard ISO wash and light tests. It was shown that stannous chloride stands out as metallic mordant, while no biomordants show sufficient intermolecular interactions to replace this metal salt. The experimental study has been corroborated with a series of molecular modeling calculations to obtain more insight into the intermolecular interactions between dye and (bio)mordants. To this end, both static Density Functional Theory based calculations as semi-empirical and force field based molecular dynamics calculations have been performed. The results indeed confirm that, in general, too small interaction energies for the biomordants of interest with the dye molecules are found, in correspondence with experimental findings. Overall, by performing systematic experiments in combination with the interpretation of the molecular models, this study yields valuable insights into the development of green routes towards use of anthocyanins as a natural dye for cellulose-based materials.
Open Access version available at UGent repository