Over the past two decades, metal–organic frameworks (MOFs) have matured from interesting academic peculiarities toward a continuously expanding class of hybrid, nanoporous materials tuned for targeted technological applications such as gas storage and heterogeneous catalysis. These oft-times crystalline materials, composed of inorganic moieties interconnected by organic ligands, can be endowed with desired structural and chemical features by judiciously functionalizing or substituting these building blocks. As a result of this reticular synthesis, MOF research is situated at the intriguing intersection between chemistry and physics, and the building block approach could pave the way toward the construction of an almost infinite number of possible crystalline structures, provided that they exhibit stability under the desired operational conditions. However, this enormous potential is largely untapped to date, as MOFs have not yet found a major breakthrough in technological applications. One of the remaining challenges for this scale-up is the densification of MOF powders, which is generally achieved by subjecting the material to a pressurization step. However, application of an external pressure may substantially alter the chemical and physical properties of the material. A reliable theoretical guidance that can presynthetically identify the most stable materials could help overcome this technological challenge.
In this Account, we describe the recent research the progress on computational characterization of the mechanical stability of MOFs. So far, three complementary approaches have been proposed, focusing on different aspects of mechanical stability: (i) the Born stability criteria, (ii) the anisotropy in mechanical moduli such as the Young and shear moduli, and (iii) the pressure-versus-volume equations of state. As these three methods are grounded in distinct computational approaches, it is expected that their accuracy and efficiency will vary. To date, however, it is unclear which set of properties are suited and reliable for a given application, as a comprehensive comparison for a broad variety of MOFs is absent, impeding the widespread use of these theoretical frameworks.
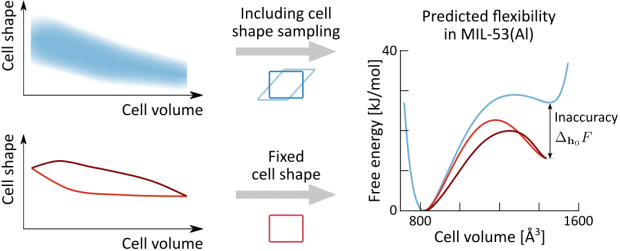
Herein, we fill this gap by critically assessing the performance of the three computational models on a broad set of MOFs that are representative for current applications. These materials encompass the mechanically rigid UiO-66(Zr) and MOF-5(Zn) as well as the flexible MIL-47(V) and MIL-53(Al), which undergo pressure-induced phase transitions. It is observed that the Born stability criteria and pressure-versus-volume equations of state give complementary insight into the macroscopic and microscopic origins of instability, respectively. However, interpretation of the Born stability criteria becomes increasingly difficult when less symmetric materials are considered. Moreover, pressure fluctuations during the simulations hamper their accuracy for flexible materials. In contrast, the pressure-versus-volume equations of state are determined in a thermodynamic ensemble specifically targeted to mitigate the effects of these instantaneous fluctuations, yielding more accurate results. The critical Account presented here paves the way toward a solid computational framework for an extensive presynthetic screening of MOFs to select those that are mechanically stable and can be postsynthetically densified before their use in targeted applications.
 Open Access version available at
Open Access version available at