Acidity effect on benzene methylation kinetics over substituted H-MeAlPO-5 catalysts
Abstract
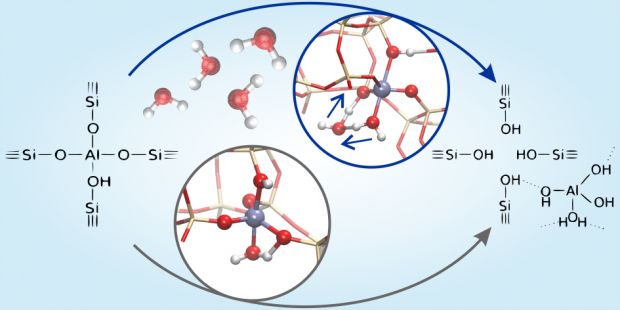
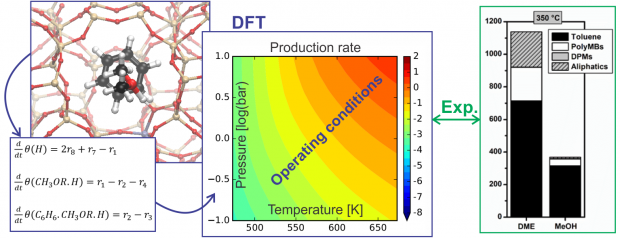
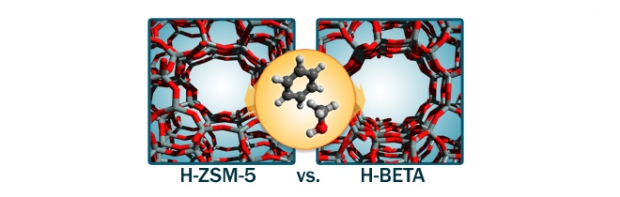
Methylation of aromatic compounds is a key reaction step in various industrial processes such as the aromatic cycle of methanol-to-hydrocarbons chemistry. The study of isolated methylation reactions and of the influence of catalyst acidity on their kinetics is a challenging task. Herein, we have studied unidirectional metal-substituted H-MeAlPO-5 materials to evaluate the effect of acid strength on the kinetics of benzene methylation with DME. First-principle simulations showed a direct correlation between the methylation barrier and acid site strength, which depends on the metal substituent. Three H-MeAlPO-5 catalysts with high (Me = Mg), moderate (Me = Si) and low acidity (Me = Zr) were experimentally tested, confirming a linear relationship between the methylation activation energy and acid strength. The effects of temperature and reactant partial pressure were evaluated, showing significant differences in the byproduct distribution between H-MgAlPO-5 and H-SAPO-5. Comparison with propene methylation suggested that the Mg substituted catalyst is also the most active for the selective methylation of alkenes.
 Open Access version available at UGent repository
Open Access version available at UGent repository