New V-IV-Based Metal-Organic Framework Having Framework Flexibility and High CO2 Adsorption Capacity
Abstract
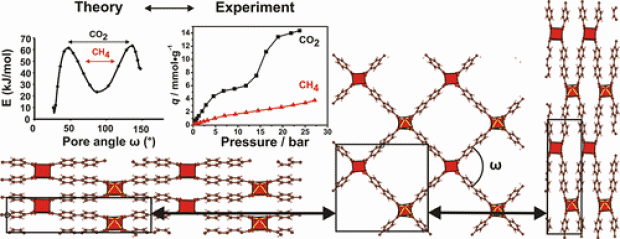
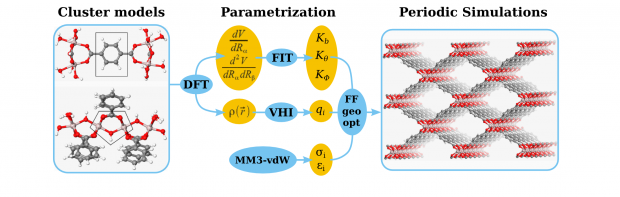
A vanadium based metal–organic framework (MOF), VO(BPDC) (BPDC2– = biphenyl-4,4′-dicarboxylate), adopting an expanded MIL-47 structure type, has been synthesized via solvothermal and microwave methods. Its structural and gas/vapor sorption properties have been studied. This compound displays a distinct breathing effect toward certain adsorptives at workable temperatures. The sorption isotherms of CO2 and CH4 indicate a different sorption behavior at specific temperatures. In situ synchrotron X-ray powder diffraction measurements and molecular simulations have been utilized to characterize the structural transition. The experimental measurements clearly suggest the existence of both narrow pore and large pore forms. A free energy profile along the pore angle was computationally determined for the empty host framework. Apart from a regular large pore and a regular narrow pore form, an overstretched narrow pore form has also been found. Additionally, a variety of spectroscopic techniques combined with N2 adsorption/desorption isotherms measured at 77 K demonstrate that the existence of the mixed oxidation states VIII/VIV in the titled MOF structure compared to pure VIV increases the difficulty in triggering the flexibility of the framework.
 Open Access version available at
Open Access version available at