On the thermodynamics of framework breathing: A free energy model for gas adsorption in MIL-53
Abstract
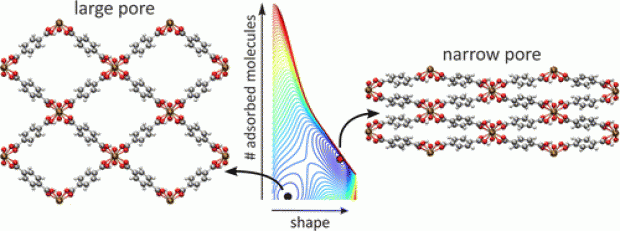
When adsorbing guest molecules, the porous metal-organic framework MIL-53(Cr) may vary its cell parameters drastically while retaining its crystallinity. A first approach to the thermodynamic analysis of this 'framework breathing' consists of comparing the osmotic potential in two distinct shapes only (large-pore and narrow-pore). In this paper, we propose a generic parametrized free energy model including three contributions: host free energy, guest-guest interactions, and host-guest interaction. Free energy landscapes may now be constructed scanning all shapes and any adsorbed amount of guest molecules. This allows to determine which shapes are the most stable states for arbitrary combinations of experimental control parameters, such as the adsorbing gas chemical potential, the external pressure, and the temperature. The new model correctly reproduces the structural transitions along the CO2 and CH4 isotherms. Moreover, our model successfully explains the adsorption versus desorption hysteresis as a consequence of the creation, stabilization, destabilization, and disappearance of a second free energy minimum under the assumptions of a first order phase transition and collective behavior. Our general thermodynamic description allows to decouple the gas chemical potential μ and mechanical pressure P as two independent thermodynamic variables and predict the complete (μ,P) phase diagram for CO2 adsorption in MIL-53(Cr). The free energy model proposed here is an important step towards a general thermodynamics description of flexible metal-organic frameworks.
 Open Access version available at
Open Access version available at