Mobility and Reactivity of Cu+ Species in Cu-CHA Catalysts under NH3-SCR-NOx Reaction Conditions: Insights from AIMD Simulations
Abstract
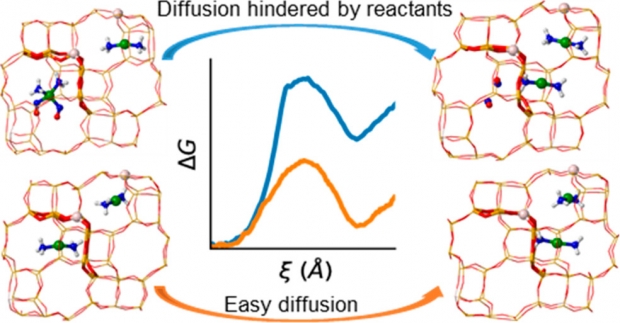
The mobility of the copper cations acting as active sites for the selective catalytic reduction of nitrogen oxides with ammonia in Cu-CHA catalysts varies with temperature and feed composition. Herein, the migration of [Cu(NH3)2]+ complexes between two adjacent cavities of the chabazite structure, including other reactant molecules (NO, O2, H2O, and NH3), in the initial and final cavities is investigated using ab initio molecular dynamics (AIMD) simulations combined with enhanced sampling techniques to describe hopping events from one cage to the other. We find that such diffusion is only significantly hindered by the presence of excess NH3 or NO in the initial cavity, since both reactants form with [Cu(NH3)2]+ stable intermediates which are too bulky to cross the 8-ring windows connecting the cavities. The presence of O2 modifies strongly the interaction of NO with Cu+. At low temperatures, we observe NO detachment from Cu+ and increased mobility of the [Cu(NH3)2]+ complex, while at high temperatures, NO reacts spontaneously with O2 to form NO2. The present simulations give evidence for recent experimental observations, namely, an NH3 inhibition effect on the SCR reaction at low temperatures, and transport limitations of NO and NH3 at high temperatures. Our first principle simulations mimicking operating conditions support the existence of two different reaction mechanisms operating at low and high temperatures, the former involving dimeric Cu(NH3)2-O2-Cu(NH3)2 species and the latter occurring by direct NO oxidation to NO2 in one single cavity.