E. Pauwels, R. Declerck, T. Verstraelen, B. De Sterck, C.W.M. Kay, V. Van Speybroeck, M. Waroquier
Journal of Physical Chemistry B
Abstract
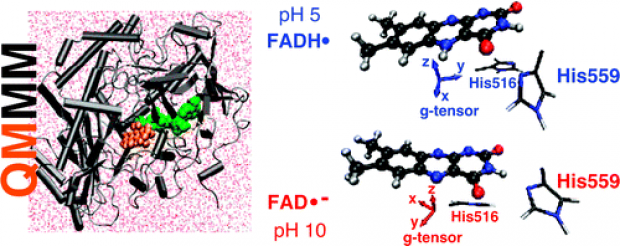
The neutral and anionic semiquinone radicals of the flavin adenine dinucleotide (FAD) cofactor noncovalently bound in glucose oxidase from A. niger are examined with the aid of QM/MM molecular modeling methods, enabling complete inclusion of the protein environment. Recently, the electron paramagnetic resonance (EPR) characteristics, the anisotropic g tensor and all the significant hyperfine couplings, of these flavoprotein radicals were determined at high resolution (J. Phys. Chem. B 2008, 112, 3568). A striking difference between the neutral and anionic radical forms was found to be a shift in the gy principal value. Within the QM/MM framework, geometry optimization and molecular dynamics simulations are combined with EPR property calculations, employing a recent implementation by some of the authors in the CP2K software package. In this way, spectroscopic characteristics are computed on the fly during the MD simulations of the solvated protein structure, mimicking as best as possible the experimental conditions. The general agreement between calculated and experimental EPR properties is satisfactory and on par with those calculated with other codes (Gaussian 03, ORCA). The protonation state of two histidines (His559 and His516) at the catalytic site of this flavoprotein is found to have a remarkable influence on the isotropic hyperfine coupling of one of the methyl groups on the neutral FAD radical, which is consistent with experimental findings in other flavoproteins (J. Biol. Chem. 2007, 282, 4738). Furthermore, the shift in the gy principal values between the neutral and anionic radicals is well reproduced by QM/MM simulations. Incorporation of at least the nearest protein environment of the cofactor radicals proves to be vital for a correct reproduction, indicating that this shift is a global feature of the protein rather than a local one. In addition, QM/MM techniques are used to make a prediction of relative angles between important spectroscopic principal directions, which are not readily determined by conventional EPR experiments. Significantly, the directions of the gx and the gy components of the g-tensor that lie in the plane of the isoalloxazine moiety are rotated by approximately 59° between the neutral and the anionic radicals.