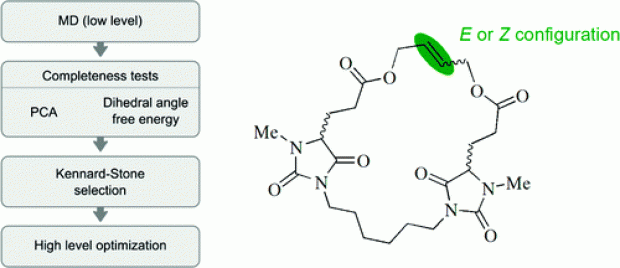
Abstract
The properties and functions of (bio)molecules are closely related to their molecular conformations. A variety of methods are available to sample the conformational space at a relatively low level of theory. If a higher level of theory is required, the computational cost can be reduced by selecting a uniformly distributed set of conformations from the ensemble of conformations generated at a low level of theory and by optimizing this selected set at a higher level. The generation of conformers is performed using molecular dynamics runs which are analyzed using the MD-Tracks code [ J. Chem. Inf. Model. 2008, 48, 2414]. This article presents a Kennard−Stone-based algorithm, with a distance measure based on the distance matrix, for the selection of the most diverse set of conformations. The method has been successfully applied to macrocyclic alkenes. The correct thermodynamic stability of the double-bond isomers of a flexible macrocyclic alkene containing two chiral centers is reproduced. The double-bond configuration has a limited effect on the conformation of the whole macrocycle. The chirality of the stereocenters has a larger effect on the molecular conformations.