
Abstract
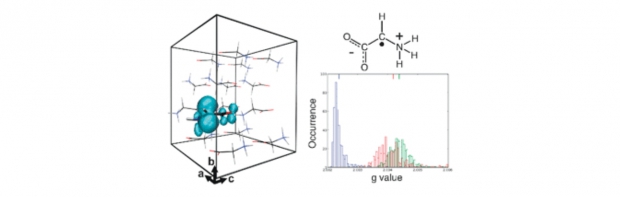
The calculation of the g tensor of the main +NH3–˙CH–COO− radiation-induced radical in solid-state α-glycine presents a real challenge to computational methods. Density functional calculations of this spectroscopic property struggle with its small anisotropy and the zwitterionic nature of the amino acids in the crystal of this seemingly simple system. Here, several factors influencing the calculated g tensor are examined by comparing with experimental data. The extent of the molecular environment is varied in both a cluster and a periodic approach and dynamic calculations are performed to account for temperature effects. The latter does not necessarily lead to a better agreement with experiment than a static calculation. Application of a periodic approach is straightforward, but an all-electron scheme clearly is favorable. In a cluster approach, the selected basis set and density functional are of less importance, provided a hybrid functional is used to prevent cluster boundary effects. The applied spin–orbit coupling operators and proper treatment of the gauge origin of the magnetic vector potential also seem to be less critical than in other, similar molecular systems. But a careful selection of the cluster size proves to be essential for this glycine radical system. The calculated g tensor varies significantly with increasing cluster size, yielding only a good agreement with experiment when 5–7 glycine molecules in the immediate environment of the central glycine radical are incorporated. Further expansion of the cluster size can even lead to an essentially incorrect description of the radical in the condensed phase, indicating that bigger clusters can become unbalanced.