Abstract
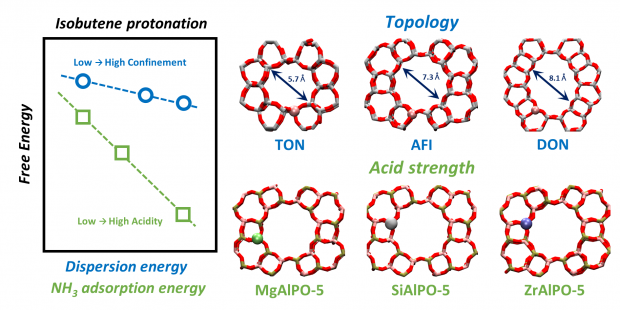
The influence of pore topology and acid strength on the adsorption of (iso)butene in Brønsted acid zeolites is investigated using a combination of static calculations and ab initio molecular dynamics simulations at operating conditions. The nature and lifetime of the adsorbed intermediates – a physisorbed alkene, a chemisorbed carbenium ion or an alkoxide – is assessed for a series of one-dimensional and three-dimensional zeolite topologies as well as metal substituted aluminophosphates with varying acid site strength. While alkoxides are elusive intermediates at high temperature, irrespective of the pore dimensions or acidity, the carbenium ion stabilization is highly correlated with the zeolite confinement and acid site strength. The impact of both topology and acidity can be nicely predicted by identifying universal descriptors such as the dispersion component of the isobutene adsorption energy (topology) and the ammonia adsorption energy (acidity). It is shown that the isobutene adsorption energies and protonation barriers follow clear linear correlations with these descriptors. Our findings yield essential insight into the reactivity differences for frameworks with a different topology and acidity. The activity of a zeolite for alkene conversion can for a large part be ascribed to variations in adsorption strength and its protonation ability.