Abstract
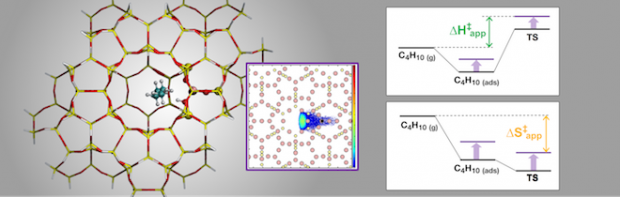
Recent experimental work has shown that variations in the confinement of n-butane at Brønsted-acid sites due to changes in zeolite framework structure strongly affect the apparent and intrinsic enthalpy and entropy of activation for cracking and dehydrogenation. Quantum chemical calculations have provided good estimates of the intrinsic enthalpies and entropies of activation extracted from experimental rate data for MFI, but extending these calculations to less confining zeolites has proven challenging, particularly for activation entropies. Herein, we report our efforts to develop a theoretical model for the cracking and dehydrogenation of n-butane occurring in a series of zeolites containing 10-membered ring channels and differing in cavity size (TON, FER, SVR, MFI, MEL, STF and MWW). We combine a QM/MM approach to calculate intrinsic and apparent activation parameters, with thermal corrections to the apparent barriers obtained from configurational-bias Monte Carlo simulations, to account for configurational contributions due to global motions of the transition state. We obtain good agreement between theory and experiment for all activation parameters for central cracking. For terminal cracking and dehydrogenation, good agreement between theory and experiment is found only at the highest confinements. Experimental activation parameters, especially those for dehydrogenation, tend to increase with decreasing confinement. This trend is not captured by the theoretical calculations, such that deviations between theory and experiment increase as confinement decreases. We propose that because transition states for dehydrogenation are later than those for cracking, relative movements between the fragments produced in the reaction become increasingly important in the less confining zeolites.