Abstract
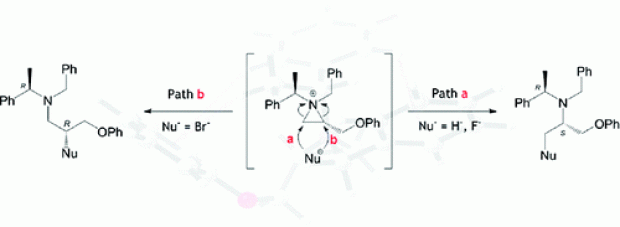
The ring opening of 2-substituted N,N-dibenzylaziridinium ions by bromide is known to occur exclusively at the substituted aziridine carbon atom via an SN2 mechanism, whereas the opposite regioselectivity has been observed as the main pathway for ring opening by fluoride. Similarly, the hydride-induced ring opening of 2-substituted N,N-dibenzylaziridinium ions has been shown to take place solely at the less hindered position. To gain insight into the main factors causing this difference in regioselectivity, a thorough and detailed computational analysis was performed on the hydride- and halide-induced ring openings of 1-benzyl-1-(α-(R)-methylbenzyl)-2(S)-(phenoxymethyl)aziridinium bromide. Intramolecular π−π stacking interactions in the aziridinium system were investigated at a range of levels that enable a proper description of dispersive interactions; a T-stacking conformer was found to be the most stable. Ring-opening mechanisms were investigated with a variety of DFT and high level ab initio methods to test the robustness of the energetics along the pathway in terms of the electronic level of theory. The necessity to utilize explicit solvent molecules to solvate halide ions was clearly shown; the potential energy surfaces for nonsolvated and solvated cases differed dramatically. It was shown that in the presence of a kinetically viable route, product distribution will be dictated by the energetically preferred pathway; this was observed in the case of hard nucleophiles (both hydride donors and fluoride). However, for the highly polarizable soft nucleophile (bromide), it was shown that in the absence of a large energy difference between transition states leading to competing pathways, the formation of the thermodynamic product is likely to be the driving force. Distortion/interaction analysis on the transition states has shown a considerable difference in interaction energies for the solvated fluoride case, pointing to the fact that sterics plays a major role in the outcome, whereas for the bromide this difference was insignificant, suggesting bromide is less influenced by the difference in sterics.
 Open Access version available at UGent repository
Open Access version available at UGent repository