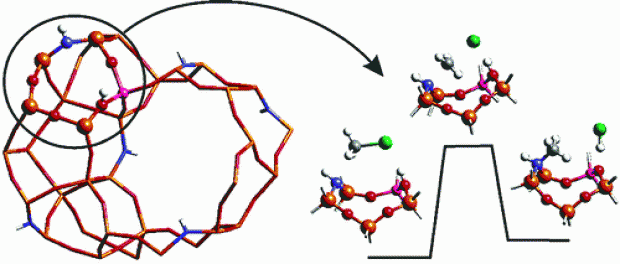
Abstract
The formation of alkylammonium groups in amine-doped zeolites is studied using density functional theory on small clusters representing the chemically active site. The presence of both strong Lewis base and Brønsted acid sites leads to a significant lowering of reaction barriers as opposed to alkoxide formation in full-oxygen zeolites. Furthermore, amine-substituted zeolites suggest novel reaction pathways that are not solely centralized around the aluminum substitution but in which two tetrahedral sites are involved, maximizing use of the zeolitic acid site and its surroundings. An investigation of the proton mobility in these yet to be synthesized materials demonstrates the need for minimizing the amount of Al−NH−Si bridges, as to prevent protonation of the amine group.