Abstract
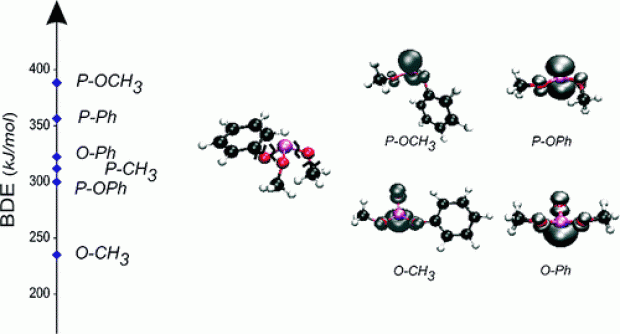
Thermodynamic properties of phosphorus-containing compounds were investigated using high-level ab initio computations. An extended set of contemporary density functional theory (DFT) procedures was assessed for their ability to accurately predict bond dissociation energies of a set of phosphoranyl radicals. The results of meta- and double-hybrids as well as more recent methods, in particular M05, M05-2X, M06, and M06-2X, were compared with benchmark G3(MP2)-RAD values. Standard heats of formation, entropies, and heat capacities of a set of ten organophosphorus compounds were determined and the low-cost BMK functional was found to provide results consistent with available experimental data. In addition, bond dissociation enthalpies (BDEs) were computed using the BMK, M05-2X, and SCS-ROMP2 procedure. The three methods give the same stability trend. The BDEs of the phosphorus(III) molecules were found to be lower than their phosphorus(V) counterparts. Overall, the following ordering is found: BDE(P−OPh)
 Open Access version available at UGent repository
Open Access version available at UGent repository