Abstract
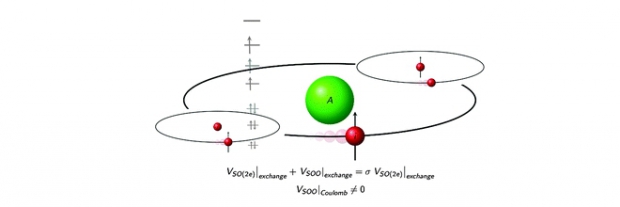
In this paper an overview is presented of several approximations within Density Functional Theory (DFT) to calculate g-tensors in transition metal containing systems and a new accurate description of the spin–other-orbit contribution for high spin systems is suggested. Various implementations in a broad variety of software packages (ORCA, ADF, Gaussian, CP2K, GIPAW and BAND) are critically assessed on various aspects including (i) non-relativistic versus relativistic Hamiltonians, (ii) spin–orbit coupling contributions and (iii) the gauge. Particular attention is given to the level of accuracy that can be achieved for codes that allow g-tensor calculations under periodic boundary conditions, as these are ideally suited to efficiently describe extended condensed-phase systems containing transition metals. In periodic codes like CP2K and GIPAW, the g-tensor calculation schemes currently suffer from an incorrect treatment of the exchange spin–orbit interaction and a deficient description of the spin–other-orbit term. In this paper a protocol is proposed, making the predictions of the exchange part to the g-tensor shift more plausible. Focus is also put on the influence of the spin–other-orbit interaction which becomes of higher importance for high-spin systems. In a revisited derivation of the various terms arising from the two-electron spin–orbit and spin–other-orbit interaction (SOO), new insight has been obtained revealing amongst other issues new terms for the SOO contribution. The periodic CP2K code has been adapted in view of this new development. One of the objectives of this study is indeed a serious enhancement of the performance of periodic codes in predicting g-tensors in transition metal containing systems at the same level of accuracy as the most advanced but time consuming spin–orbit mean-field approach. The methods are first applied on rhodium carbide but afterwards extended to a broad test set of molecules containing transition metals from the fourth, fifth and sixth row of the periodic table. The set contains doublets as well as high-spin molecules.