Thermodynamic insight into stimuli-responsive behavior of soft porous crystals
Abstract
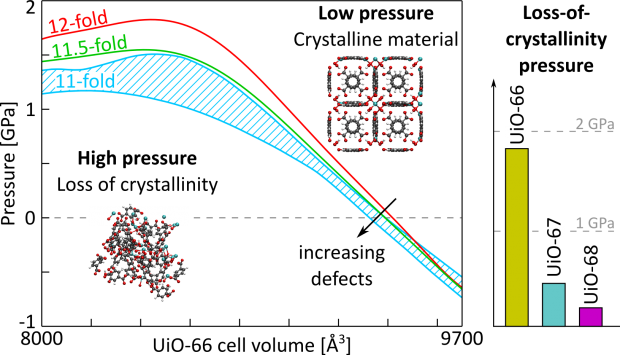
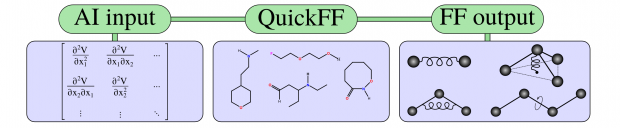
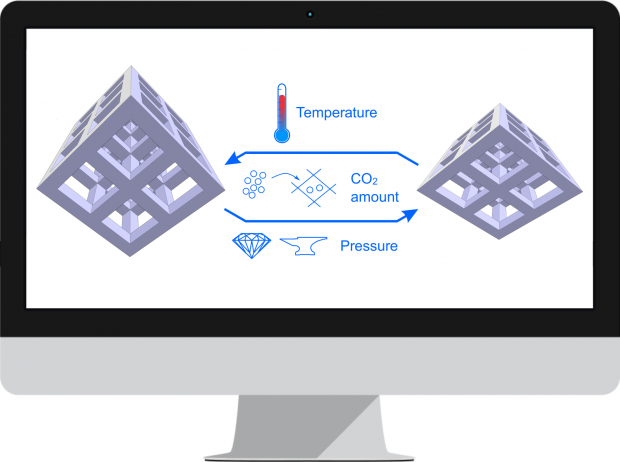
Knowledge of the thermodynamic potential in terms of the independent variables allows to characterize the macroscopic state of the system. However, in practice, it is difficult to access this potential experimentally due to irreversible transitions that occur between equilibrium states. A showcase example of sudden transitions between (meta)stable equilibrium states is observed for soft porous crystals possessing a network with long-range structural order, which can transform between various states upon external stimuli such as pressure, temperature and guest adsorption. Such phase transformations are typically characterized by large volume changes and may be followed experimentally by monitoring the volume change in terms of certain external triggers. Herein, we present a generalized thermodynamic approach to construct the underlying Helmholtz free energy as a function of the state variables that governs the observed behaviour based on microscopic simulations. This concept allows a unique identification of the conditions under which a material becomes flexible.
 Open Access version available at UGent repository
Open Access version available at UGent repository