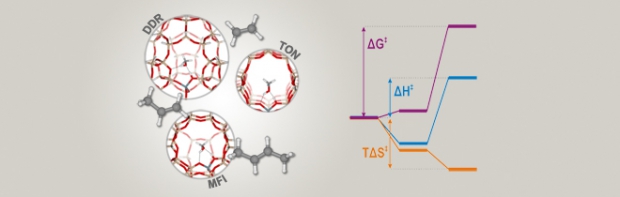
Enthalpy and entropy barriers explain the effects of topology on the kinetics of zeolite-catalyzed reactions

Abstract
The methylation of ethene, propene, and trans-2-butene on zeolites H-ZSM-58 (DDR), H-ZSM-22 (TON), and H-ZSM-5 (MFI) is studied to elucidate the particular influence of topology on the kinetics of zeolite-catalyzed reactions. H-ZSM-58 and H-ZSM-22 are found to display overall lower methylation rates compared to H-ZSM-5 and also different trends in methylation rates with increasing alkene size. These variations may be rationalized based on a decomposition of the free-energy barriers into enthalpic and entropic contributions, which reveals that the lower methylation rates on H-ZSM-58 and H-ZSM-22 have virtually opposite reasons. On H-ZSM-58, the lower methylation rates are caused by higher enthalpy barriers, owing to inefficient stabilization of the reaction intermediates in the large cage-like pores. On the other hand, on H-ZSM-22, the methylation rates mostly suffer from higher entropy barriers, because excessive entropy losses are incurred inside the narrow-channel structure. These results show that the kinetics of crucial elementary steps hinge on the balance between proper stabilization of the reaction intermediates inside the zeolite pores and the resulting entropy losses. These fundamental insights into their inner workings are indispensable for ultimately selecting or designing better zeolite catalysts.
 Open Access version available at
Open Access version available at