Synthesis, characterization and sorption properties of NH2-MIL-47
Abstract
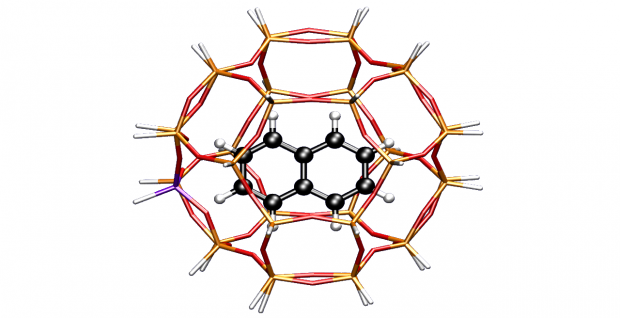
An amino functionalized vanadium-containing Metal Organic Framework, NH2-MIL-47 has been synthesized by a hydrothermal reaction in an autoclave. Alternatively, a synthesis route via microwave enhanced irradiation has been optimized to accelerate the synthesis. The NH2-MIL-47 exhibits the same topology as MIL-47, in which the V center is octahedrally coordinated. After an exchange procedure in DMF the V+III center is oxidized to V+IV, which is confirmed by EPR and XPS measurements. The CO2 and CH4 adsorption properties have been evaluated and compared to MIL-47, showing that both MOFs have an almost similar adsorption capacity and affinity for CO2. DFT- based molecular modeling calculations were performed to obtain more insight into the adsorption positions for CO2 in NH2-MIL-47. Furthermore our calculated adsorption enthalpies agree well with the experimental values.
 Open Access version available at
Open Access version available at