Modeling Gas Adsorption in Flexible Metal–Organic Frameworks via Hybrid Monte Carlo / Molecular Dynamics Schemes
Abstract
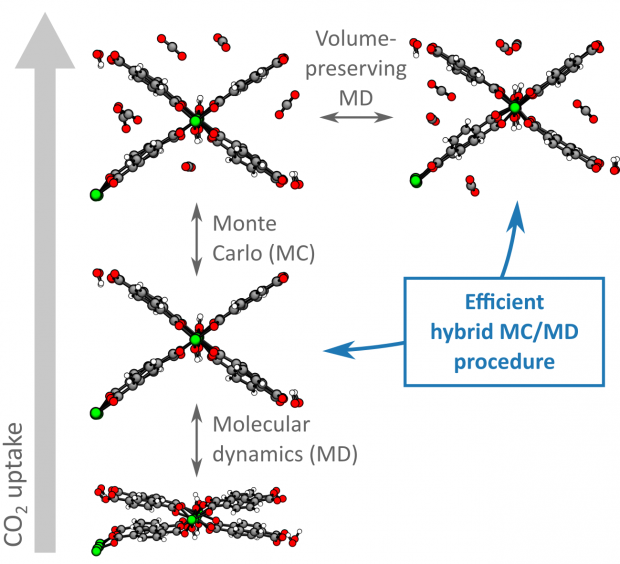
Herein, a hybrid Monte Carlo (MC)/molecular dynamics (MD) simulation protocol that properly accounts for the extraordinary structural flexibility of metal–organic frameworks (MOFs) is developed and validated. This is vital to accurately predict gas adsorption isotherms and guest‐induced flexibility of these materials. First, the performance of three recent models to predict adsorption isotherms and flexibility in MOFs is critically investigated. While these methods succeed in providing qualitative insight in the gas adsorption process in MOFs, their accuracy remains limited as the intrinsic flexibility of these materials is very hard to account for. To overcome this challenge, a hybrid MC/MD simulation protocol that is specifically designed to handle the flexibility of the adsorbent, including the shape flexibility, is introduced, thereby unifying the strengths of the previous models. It is demonstrated that the application of this new protocol to the adsorption of neon, argon, xenon, methane, and carbon dioxide in MIL‐53(Al), a prototypical flexible MOF, substantially decreases the inaccuracy of the obtained adsorption isotherms and predicted guest‐induced flexibility. As a result, this method is ideally suited to rationalize the adsorption performance of flexible nanoporous materials at the molecular level, paving the way for the conscious design of MOFs as industrial adsorbents.