Modeling the structural and thermal properties of loaded metal-organic frameworks. An interplay of quantum and anharmonic fluctuations
Journal of Chemical Theory and Computation
15 (5), 3237-3249
2019
A1
In this article, we investigate the influence of anharmonicities and nuclear quantum effects (NQEs) in modelling the structural properties and thermal expansion of the empty MOF-5 metal-organic framework. To introduce NQEs in classical molecular dynamics simulations, two different methodologies are considered, comparing the approximate, but computationally cheap, method of generalised Langevin equation thermostatting to the more advanced, computationally demanding path integral molecular dynamics technique. For both methodologies, similar results were obtained for all the properties under investigation. The structural properties of MOF-5, probed by means of radial distribution functions (RDFs), show some distinct differences with respect to a classical description. Besides a broadening of the RDF peaks under the influence of quantum fluctuations, a different temperature dependence is also observed due to a dominant zero-point energy (ZPE) contribution. For the thermal expansion of MOF-5, by contrast, NQEs appear to be only of secondary importance with respect to an adequate modelling of the anharmonicities of the potential energy surface (PES), as demonstrated by the use of two differently parametrised force fields. Despite the small effect in the temperature dependence of the volume of MOF-5, NQEs do however significantly affect the absolute volume of MOF-5, in which the ZPE resulting from the intertwining of NQEs and anharmonicities plays a crucial role. A sufficiently accurate description of the PES is therefore prerequisite when modelling NQEs.
The mechanism inducing the breathing in flexible metal-organic frameworks, such as MIL-53(Al), is still not fully understood. Herein, the influence of lattice vibrations on the breathing transition in MIL-53(Al) is investigated to gain insight in this phenomenon. Through solid-state density-functional theory calculations, the volume-dependent IR spectrum is computed together with the volume-frequency relations of all vibrational modes. Furthermore, important thermodynamic properties such as the Helmholtz free energy, the specific heat capacity, the bulk modulus, and the volumetric thermal expansion coefficient are derived via these volume-frequency relations using the quasi-harmonic approximation. The simulations expose a general volume-dependency of the vibrations with wavenumbers above 300 cm−1 due to their localized nature. In contrast, a diverse set of volume-frequency relations are observed for vibrations in the terahertz region (< 300 cm−1) containing the vibrations exhibiting collective behavior. Some terahertz vibrations display large frequency differences over the computed volume range, induced by either repulsion or strain effects, potentially triggering the phase transformation. Finally, the impact of the lattice vibrations on the thermodynamic properties is investigated. This reveals that the closed pore to large pore phase transformation in MIL-53(Al) is mainly facilitated by terahertz vibrations inducing rotations of the organic linker, while the large pore to closed pore phase transformation relies on two framework-specific soft modes.
Various kinds of flexibility have been observed in metal–organic frameworks, which may originate from the topology of the material or the presence of flexible ligands. The construction of free energy profiles describing the full dynamical behavior along the phase transition path is challenging since it is not trivial to identify collective variables able to identify all metastable states along the reaction path. In this work, a systematic three-step protocol to uniquely identify the dominant order parameters for structural transformations in flexible metal–organic frameworks and subsequently construct accurate free energy profiles is presented. Methodologically, this protocol is rooted in the time-structure based independent component analysis (tICA), a well-established statistical modeling technique embedded in the Markov state model methodology and often employed to study protein folding, that allows for the identification of the slowest order parameters characterizing the structural transformation. To ensure an unbiased and systematic identification of these order parameters, the tICA decomposition is performed based on information from a prior replica exchange (RE) simulation, as this technique enhances the sampling along all degrees of freedom of the system simultaneously. From this simulation, the tICA procedure extracts the order parameters—often structural parameters—that characterize the slowest transformations in the material. Subsequently, these order parameters are adopted in traditional enhanced sampling methods such as umbrella sampling, thermodynamic integration, and variationally enhanced sampling to construct accurate free energy profiles capturing the flexibility in these nanoporous materials. In this work, the applicability of this tICA-RE protocol is demonstrated by determining the slowest order parameters in both MIL-53(Al) and CAU-13, which exhibit a strongly different type of flexibility. The obtained free energy profiles as a function of this extracted order parameter are furthermore compared to the profiles obtained when adopting less-suited collective variables, indicating the importance of systematically selecting the relevant order parameters to construct accurate free energy profiles for flexible metal–organic frameworks, which is in correspondence with experimental findings. The method succeeds in mapping the full free energy surface in terms of appropriate collective variables for MOFs exhibiting linker flexibility. For CAU-13, we show the decreased stability of the closed pore phase by systematically adding adsorbed xylene molecules in the framework.
Progress in the atomic-scale modelling of matter over the past decade has been tremendous. This progress has been brought about by improvements in methods for evaluating interatomic forces that work by either solving the electronic structure problem explicitly, or by computing accurate approximations of the solution and by the development of techniques that use the Born-Oppenheimer (BO) forces to move the atoms on the BO potential energy surface. As a consequence of these developments it is now possible to identify stable or metastable states, to sample configurations consistent with the appropriate thermodynamic ensemble, and to estimate the kinetics of reactions and phase transitions. All too often, however, progress is slowed down by the bottleneck associated with implementing new optimization algorithms and/or sampling techniques into the many existing electronic-structure and empirical-potential codes. To address this problem, we are thus releasing a new version of the i-PI software. This piece of software is an easily extensible framework for implementing advanced atomistic simulation techniques using interatomic potentials and forces calculated by an external driver code. While the original version of the code was developed with a focus on path integral molecular dynamics techniques, this second release of i-PI not only includes several new advanced path integral methods, but also offers other classes of algorithms. In other words, i-PI is moving towards becoming a universal force engine that is both modular and tightly coupled to the driver codes that evaluate the potential energy surface and its derivatives.
 Open Access version available at UGent repository
Open Access version available at UGent repository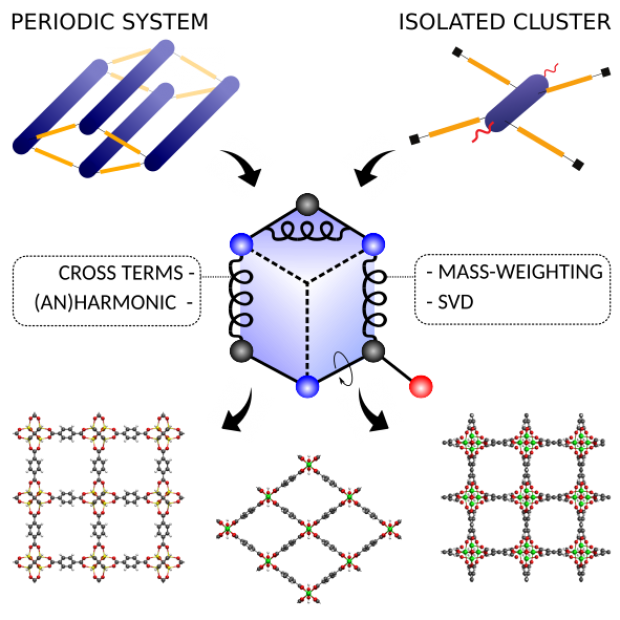
QuickFF was originally launched in 2015 to derive accurate force fields for isolated and complex molecular systems in a quick and easy way. Apart from the general applicability, the functionality was especially tested for metal-organic frameworks (MOFs), a class of hybrid materials consisting of organic and inorganic building blocks. Herein, we launch a new release of the QuickFF protocol which includes new major features to predict structural, vibrational, mechanical and thermal properties with greater accuracy, without compromising its robustness and transparant workflow. First, the ab initio data necessary for the fitting procedure may now also be derived from periodic models for the molecular system, as opposed to the earlier cluster-based models. This is essential for an accurate description of MOFs with one dimensional metal-oxide chains. Second, cross terms that couple internal coordinates (ICs) and anharmonic contributions for bond and bend terms are implemented. These features are essential for a proper description of vibrational and thermal properties. Third, the fitting scheme was modified to improve robustness and accuracy. The new features are tested on MIL-53(Al), MOF-5, CAU-13 and NOTT-300. As expected, periodic input data is proven to be essential for a correct description of structural, vibrational and thermodynamic properties of MIL-53(Al). Bulk moduli and thermal expansion coefficients of MOF-5 are very accurately reproduced by static and dynamic simulations using the newly derived force fields which include cross terms and anharmonic corrections. For the flexible materials CAU-13 and NOTT-300, the transition pressure is accurately
predicted provided cross terms are taken into account.
Open Access version available at UGent repository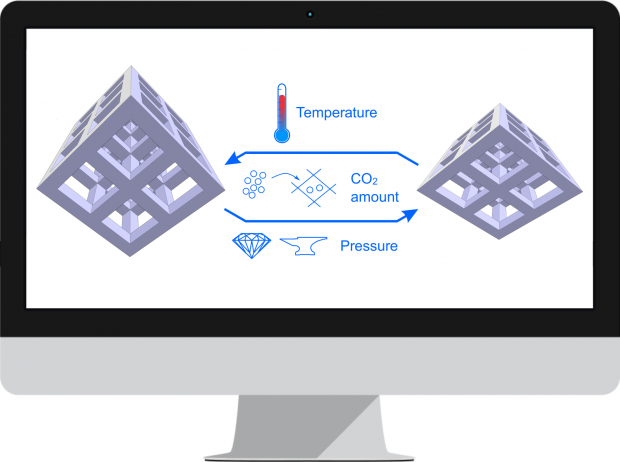
Knowledge of the thermodynamic potential in terms of the independent variables allows to characterize the macroscopic state of the system. However, in practice, it is difficult to access this potential experimentally due to irreversible transitions that occur between equilibrium states. A showcase example of sudden transitions between (meta)stable equilibrium states is observed for soft porous crystals possessing a network with long-range structural order, which can transform between various states upon external stimuli such as pressure, temperature and guest adsorption. Such phase transformations are typically characterized by large volume changes and may be followed experimentally by monitoring the volume change in terms of certain external triggers. Herein, we present a generalized thermodynamic approach to construct the underlying Helmholtz free energy as a function of the state variables that governs the observed behaviour based on microscopic simulations. This concept allows a unique identification of the conditions under which a material becomes flexible.
Open Access version available at UGent repository
In this work mid-infrared (mid-IR), far-IR, and Raman spectra are presented for the distinct (meta)stable phases of the flexible metal-organic framework MIL-53(Al). Static density functional theory (DFT) simulations are performed allowing for the identification of all IR active modes, which is unprecedented in the low-frequency region. A unique vibrational fingerprint is revealed, resulting from aluminum-oxide backbone stretching modes, which can be used to clearly distinguish the IR spectra of the closed- and large-pore phases. Furthermore, molecular dynamics simulations based on a DFT description of the potential energy surface enable to determine the theoretical Raman spectrum of the closed- and large-pore phases for the first time. An excellent correspondence between theory and experiment is observed. Both the low-frequency IR and Raman spectra show major differences in vibrational modes between the closed- and large-pore phases indicating changes in lattice dynamics between the two structures. In addition, several collective modes related to the breathing mechanism in MIL-53(Al) are identified. In particular, we rationalize the importance of the trampoline-like motion of the linker for the phase transition.
Open Access version available at UGent repositoryIn order to reliably predict and understand the breathing behavior of highly flexible metal–organic frameworks from thermodynamic considerations, an accurate estimation of the free energy difference between their different metastable states is a prerequisite. Herein, a variety of free energy estimation methods are thoroughly tested for their ability to construct the free energy profile as a function of the unit cell volume of MIL-53(Al). The methods comprise free energy perturbation, thermodynamic integration, umbrella sampling, metadynamics, and variationally enhanced sampling. A series of molecular dynamics simulations have been performed in the frame of each of the five methods to describe structural transformations in flexible materials with the volume as the collective variable, which offers a unique opportunity to assess their computational efficiency. Subsequently, the most efficient method, umbrella sampling, is used to construct an accurate free energy profile at different temperatures for MIL-53(Al) from first principles at the PBE+D3(BJ) level of theory. This study yields insight into the importance of the different aspects such as entropy contributions and anharmonic contributions on the resulting free energy profile. As such, this thorough study provides unparalleled insight in the thermodynamics of the large structural deformations of flexible materials.
Open Access version available at UGent repository