K. Hendrickx
A series of sulfonic acid functionalized mixed-linker DUT-4 analogues: synthesis, gas sorption properties and catalytic performance
Dalton Transactions
46, 14356
2017
A1
Abstract
In this work, we present the successful synthesis of a series of sulfonic acid functionalized mixed-linker metal–organic frameworks (MOFs) having the DUT-4 topology by using different ratios of 2,6-naphthalenedicarboxylic acid (H2-NDC) and 4,8-disulfonaphthalene-2,6-dicarboxylic acid (H2-NDC-2SO3H) in one-pot reactions. The obtained materials were fully characterized and their CO2 adsorption properties at low and high pressures were studied and compared with those of the pristine DUT-4 material. Generally, the CO2 adsorption capacities range from 3.28 and 1.36 mmol g−1 for DUT-4 to 1.54 and 0.78 mmol g−1 for DUT-4-SO3H (50) up to 1 bar at 273 K and 303 K, respectively. Computational calculations corroborated the structural changes of the material in function of the loading of sulfonic acid groups. Furthermore, due to the strong Brønsted acid character, the resulting sulfonic acid based MOF material was evaluated as a catalyst for the ring opening of styrene oxide with methanol as a nucleophile under mild conditions, showing almost full conversion (99%) after 5 hours of reaction. A hot filtration experiment demonstrated that the catalysis occurred heterogeneously and the catalyst could be recovered and reused for multiple runs without significant loss in activity and crystallinity.
Missing linkers: an alternative pathway to UiO-66 electronic structure engineering
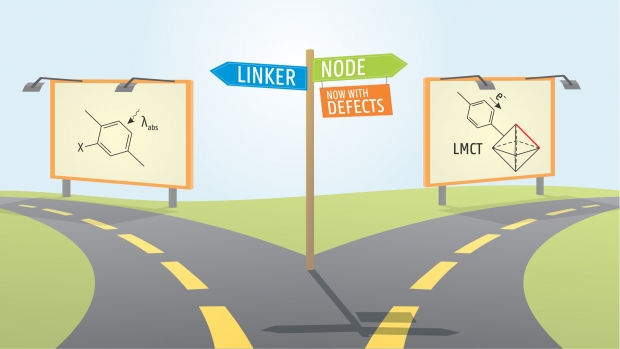
Chemistry of Materials
29 (7), 3006–3019
2017
A1
Abstract
UiO-66 is a promising metal-organic framework for photocatalytic applications. However, the ligand-to-metal charge transfer of an excited electron is inefficient in the pristine material. Herein we assess the influence of missing linker defects on the electronic structure of UiO-66 and discuss their ability to improve ligand-to-metal charge transfer. Using a new defect classification system, which is transparent and easily extendable, we identify the most promising photocatalysts by considering both relative stability and electronic structure. We find the properties of UiO-66 defect structures largely to depend on the coordination of the constituent nodes, and the nodes with the strongest local distortions to alter the electronic structure most. Defects hence provide an alternative pathway to tune UiO-66 for photocatalytic purposes, besides linker modification and node metal substitution. In addition, the decomposition of MOF properties into node- and linker-based behavior is more generally valid, so we propose orthogonal electronic structure tuning as a paradigm in MOF electronic structure engineering.
 Open Access version available at UGent repository
Open Access version available at UGent repositoryGold Open Access
Heterogeneous Ru(III) oxidation catalysts via ‘click’ bidentate ligands on a Periodic Mesoporous Organosilica support
Green Chemistry
18, 6035–6045
2016
A1
Abstract
A 100% monoallyl ring-type Periodic Mesoporous Organosilica (PMO) is prepared as a novel, versatile and exceptionally stable catalytic support with a high internal surface area and 5.0 nm pores. Thiol-ene ‘click’ chemistry allows straightforward attachment of bifunctional thiols (-NH2, -OH, -SH) which, exploiting the thioether functionality formed, give rise to ‘solid’ bidentate ligands. [Ru(acac)2(CH3CN)2]PF6 is attached and complex formation on the solid is studied via theoretical calculations. All resulting solid catalysts show high activity and selectivity in alcohol oxidation reactions performed in green conditions (25°C/ water). The PMO catalysts do not leach Ru during reaction and are thus easily recuperated and re-used for several runs. Furthermore, oxidation of poorly water-soluble (±)-menthol illustrates the benefits of using hydrophobic PMOs as catalytic supports.
Facile synthesis of cooperative acid-base catalysts by clicking cysteine and cysteamine on an ethylene-bridged periodic mesoporous organosilica
European Journal of Inorganic Chemistry
2016, 13-14, 2144-2151
2016
A1
Abstract
A Periodic Mesoporous Organosilica (PMO) containing ethylene bridges was functionalized in order to obtain a series of cooperative acid-base catalysts. A straightforward, single-step procedure was devised to immobilize cysteine and cysteamine on the support material via a photoinitiated thiol-ene click reaction. Likewise, PMO materials capped with hexamethyldisilazane (HMDS) were used to support both compounds. This resulted in different materials, where the amine site was promoted by carboxylic acid groups, surface silanol groups or both. The catalysts were tested in the aldol reaction of 4-nitrobenzaldehyde and acetone. It was found that silanol groups have a stronger promoting effect on the amine than the carboxylic acid group. The highest turnover frequency (TOF) was obtained for an amine functionalized material which only contained silanol promoting sites. The loading of the active sites also has a significant effect on the activity of the catalysts, which was rationalized based on a computational study.
Systematic study of the chemical and hydrothermal stability of selected "stable" Metal Organic Frameworks
Microporous and Mesoporous Materials
226, 110-116
2016
A1
Abstract
In this work, the hydrothermal and chemical stability towards acids, bases, air, water and peroxides of Metal Organic Frameworks, that are commonly considered to be stable, is presented. As a proof of stability both the crystallinity and porosity are measured before and after exposure to the stress test. The major part of the MOFs examined in this study showed a good hydrothermal stability except for the UiO-67, NH2-MIL-101 (Al) and CuBTC material. The chemical stability towards acids and bases show a similar tendency and an ordering can be proposed as: MIL-101(Cr)>NH2-UiO-66>UiO-66>UiO-67>NH2-MIL-53>MIL-53(Al)>ZIF-8>CuBTC>NH2-MIL-101(Al). In the tests with the H2O2 solution most materials behaved poorly, only the UiO-66 and NH2-UiO-66 framework showed a good stability.
Vibrational fingerprint of the absorption properties of UiO-type MOF materials
Theoretical Chemistry Accounts
135, 4, 102
2016
A1
Abstract
The absorption properties of UiO-type metal–organic frameworks are computed using TD-DFT simulations on the organic linkers. A set of nine isoreticular structures, including the UiO-66 and UiO-67 materials and functionalized variants, are examined. The excitation energies from a static geometry optimization are compared with dynamic averages obtained from sampling the ground-state potential energy surface using molecular dynamics. The vibrational modes that impact the excitation energy are identified. This analysis is done using a recently proposed tool based on power spectra of the velocities and the excitation energies. The applied procedure allows including important factors influencing the absorption spectra, such as the periodic framework, linker variation and dynamical effects including harmonic and anharmonic nuclear motions. This methodology allows investigating in detail the vibrational fingerprint of the excitation energy of advanced materials such as MOFs and gives perspectives to tailor materials toward new light-based applications.
Open Access version available at UGent repositoryUnderstanding Intrinsic Light Absorption Properties of UiO- 66 Frameworks: A Combined Theoretical and Experimental Study
Inorganic Chemistry
54, 22, 10701-10710
2015
A1
Abstract
A combined theoretical and experimental study is performed in order to elucidate the effects of linker functional groups on the photoabsorption properties of UiO-66-type materials. This study, in which both mono- and di-functionalized linkers (with X= -OH, -NH2, -SH) are studied, aims to obtain a more complete picture on the choice of functionalization. Static Time-Dependent Density Functional Theory (TD-DFT) calculations combined with Molecular Dynamics simulations are performed on the linkers and compared to experimental UV/VIS spectra, in order to understand the electronic effects governing the absorption spectra. Di-substituted linkers show larger shifts compared to mono-substituted variants, making them promising candidates for further study as photocatalysts. Next, the interaction between the linker and the inorganic part of the framework is theoretically investigated using a cluster model. The proposed Ligand-to-Metal-Charge Transfer (LMCT) is theoretically observed and is influenced by the differences in functionalization. Finally, computed electronic properties of the periodic UiO-66 materials reveal that the band gap can be altered by linker functionalization and ranges from 4.0 down to 2.2 eV. Study of the periodic Density of States (DOS) allows to explain the band gap modulations of the framework in terms of a functionalization-induced band in the band gap of the original UiO-66 host.
More insight in multiple bonding with valence bond theory
Computational and Theoretical Chemistry
1053, pp. 180-188
2015
A1
Published while none of the authors were employed at the CMM
Abstract
An original procedure is proposed, based on valence bond theory, to calculate accurate dissociation energies for multiply bonded molecules, while always dealing with extremely compact wave functions involving three valence bond structures at most. The procedure consists of dividing the bond-breaking into sequential steps, thus breaking one by one the separate components of the multiple bond. By using the breathing-orbital valence bond method (Hiberty and Shaik, 2002), it is ensured that both static and dynamic differential electron correlations are taken into account in each step. The procedure is illustrated for typical examples of multiply bonded molecules, N2, C2 and CO. The so-calculated total dissociation energies are at par with accurate calculations by state-of-the-art standard methods in the same basis set. The procedure also allows one to get some deep insight into the properties of the individual bonds that constitute the multiple bond. A so-called quasi-classical state is defined, in which the electrons of the bond under study have only one spin arrangement pattern, αβ, thus disabling the exchange of the two spin arrangements that is necessary for a covalent bonding interaction to take place. Taking this quasi-classical state as a non-bonded reference, one may estimate the “in-situ bonding energy” of an individual bond, as calculated at the molecular equilibrium geometry and in the presence of the other electrons. The procedure may also be used to assess the preferred bond length of an individual bond, which is shown to amount to 1.33 Å for the σ bond of N2, while the π bonds get stronger and stronger as the interatomic distance is shortened. Another application is the calculation of the resonance energy arising from the mixing of the ionic components of an individual bond to its covalent component, and the comparison of this resonance energy with the in-situ bonding energy. This shows that the σ bond of N2 and C2 is a classical covalent bond. On the other hand, the π bonds have a substantial resonance energy that put them close to the category of charge-shift bonds.
Multicenter Bonding in Ditetracyanoethylene Dianion: A Simple Aromatic Picture in Terms of Three-Electron Bonds
Journal of Chemical Theory and Computation (JCTC)
9 (5), 2276–2285
2013
A1
Published while none of the authors were employed at the CMM
Abstract
The nature of the multicenter, long bond in ditetracyanoethylene dianion complex [TCNE]22– is elucidated using high level ab initio Valence Bond (VB) theory coupled with Quantum Monte Carlo (QMC) methods. This dimer is the prototype of the general family of pancake-bonded dimers with large interplanar separations. Quantitative results obtained with a compact wave function in terms of only six VB structures match the reference CCSD(T) bonding energies. Analysis of the VB wave function shows that the weights of the VB structures are not compatible with a covalent bond between the π* orbitals of the fragments. On the other hand, these weights are consistent with a simple picture in terms of two resonating bonding schemes, one displaying a pair of interfragment three-electron σ bonds and the other displaying intrafragment three-electron π bonds. This simple picture explains at once (1) the long interfragment bond length, which is independent of the countercations but typical of three-electron (3-e) CC σ bonds, (2) the interfragment orbital overlaps which are very close to the theoretical optimal overlap of 1/6 for a 3-e σ bond, and (3) the unusual importance of dynamic correlation, which is precisely the main bonding component of 3-e bonds. Moreover, it is shown that the [TCNE]22– system is topologically equivalent to the square C4H42– dianion, a well-established aromatic system. To better understand the role of the cyano substituents, the unsubstituted diethylenic Na+2[C2H4]22– complex is studied and shown to be only metastable and topologically equivalent to a rectangular C4H42– dianion, devoid of aromaticity.