The effect of confined space on the growth of naphthalenic species in an H-SSZ-13 catalyst: a molecular modeling study
ChemCatChem
1 (3), 373-378
2009
A1

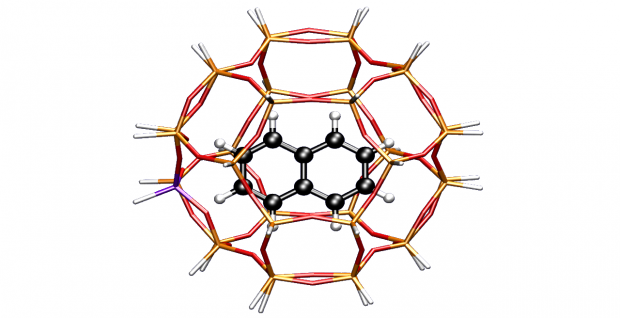
Abstract
Methylation reactions of naphthalenic species over the acidic microporous zeolite with chabazite topology have been investigated by means of two-layered ab initio computations. Large cluster results combined with van der Waals contributions provide thermodynamic and kinetic results of successive methylation steps. The growth of fused bicyclic species is important as these can act as hydrocarbon pool species within the methanol-to-olefin (MTO) process, but ultimately leads to the deactivation of the catalyst. The influence of the confined space of the zeolite pore on the resulting transition state or product shape selectivity is investigated in detail.