Together with CNRS Montpellier we are looking for two Joint-PhD students within the frame of the Marie-Skłodowska-Curie Doctoral Network SENNET within the Horizon Europe Programme of the European Commission. SENNET is the “Porous Networks for Gas Sensing” project and will create disruptive sensor technology for indoor air quality by incorporating porous materials and sensor technology. The project has pooled the interdisciplinary and intersectoral expertise of leading members located in Belgium, Germany, France, Ireland, Moldova, Spain, UK and the Netherlands (Figure 1). The 12 SENNET researchers will not only receive state-of-the-art science/technology training but will also benefit from a unique soft-skills training programme. This will kick-start their careers as highly employable professionals in the EU and beyond.
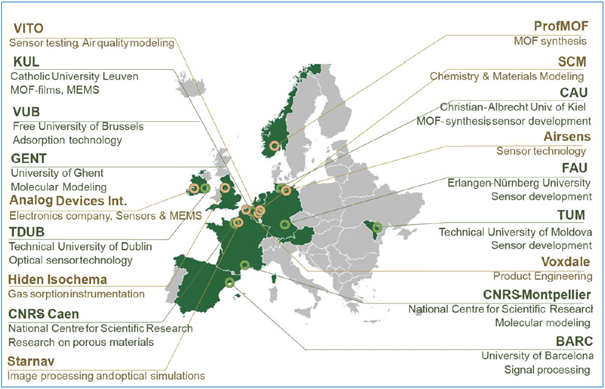
Figure 1 - SENNET Consortium
About the project
Air pollution is one of the most pressing environmental challenges worldwide. While outdoor air pollution appears often in the media, the effects of indoor air pollution are not to be underestimated since the average person spends about 22 h per day indoors. Many chemicals that decrease the quality of indoor air are emitted by carpets, paints, furniture, etc. The majority of these pollutants are volatile organic compounds (VOCs). Since VOCs can cause not only discomfort but also debilitating or even fatal conditions, it is desirable to measure VOC concentrations with high spatial and temporal resolution, via low-cost but reliable miniature sensors. However, selectively measuring harmful VOCs is particularly challenging because of the low concentration of the analyte and the multitude of interfering compounds present in indoor air. Currently available miniature sensors (e.g., metal oxide semiconductor sensors) typically cannot distinguish a VOC of interest from, for instance, an air freshener.
SENNET will develop novel sensors for the selective detection of priority VOCs, based on leveraging the adsorption properties of tunable porous materials, namely metal-organic frameworks (MOFs) and zeolites. To do so, we will bridge the gap between two fields, namely porous crystalline materials and sensor technology, that have thus far been separated by traditional subject boundaries. SENNET is the first training network that will tackle this challenge, and will do so by combining expertise in chemistry, physics, materials engineering, and sensors. A coordinated effort by 9 beneficiaries and 7 associated partner organizations from 8 countries guarantees a pan-European approach in a multi-environment context (universities, research centers, SMEs and large companies). The proposed ‘follow-the-challenge’ strategy ensures that young researchers are exposed to a variety of research environments and get involved in each step of the materials & sensors value chain.
Breakthrough materials for developing these novel sensors are MOFs and zeolites, crystalline, porous network materials. Whereas zeolites are inorganic aluminosilicates containing extra-framework cations, MOFs are hybrid solids that consist of inorganic nodes connected by multitopic organic molecules. Both classes of material have uniform pores with dimensions comparable to the size of the VOCs to be detected and have high surface areas (up to 6000 m2 g-1). Because of their chemical and structural characteristics, MOFs and zeolites can capture VOCs even at trace concentrations. Moreover, the adsorption preference (or selectivity) can be tuned by changing the nature of the framework. Although these adsorption properties are potentially very promising for VOC sensors, the integration of MOFs or zeolites into real devices has been largely overlooked. Firstly, due to a lack of systematic knowledge about which of the many different MOFs or zeolites would be best suited to adsorb a particular VOC from the air, and, secondly, the lack of suitable methods to integrate these materials with a sensor technology, e.g., coating deposition and signal transduction.
SENNET will address these challenges by combining expertise from fundamental chemistry all the way through to sensor engineering, resulting in the scientific objectives (SOs) outlined below . The overall approach is to combine multiple sensor elements coated by a porous material that displays cross-selective but different adsorption behaviour. The nature and concentration of the target VOC will be determined from the combined response of all the sensor elements through a multivariate calibration approach borrowed from chemometrics. The focus will be on the priority risk VOCs determined by the World Health Organization, i.e., tetrachloroethylene, formaldehyde and benzene.
The project’s five Scientific Objectives (SOs) are defined below:
- Because of the large numbers of MOFs (> 10,000) and zeolites (> 240), and their numerous multicomponent adsorption behaviours, an efficient screening approach is needed. High-throughput computational approaches will be developed to tackle this problem and identify structure-adsorption property relationships.
- The predicted behaviour must be validated by experimental adsorption tests for both single components and complex mixtures to fully understand the behaviour of the materials in indoor air.
- Synthesis of MOFs and zeolites and fine-tuning of their properties to obtain the required mixed-component adsorption behaviour, based on the identified structure-adsorption property relationships. The materials will be prepared in a form that facilitates their integration in sensors.
- Sensor fabrication and testing based on the most promising MOFs and zeolites. New fabrication approaches will be explored and the testing will involve real-world conditions. Different strategies to transduce the VOCs’ adsorption characteristics into a measurable signal will be benchmarked.
- Signal-processing methods will be developed to calibrate the sensors and sensor arrays and to prevent non-selective or drifting signals as a result of temperature, relative humidity, or interfering compounds.
About Ghent University and CMM
Ghent University is a top 100 university and one of the major universities in Belgium. Five Doctoral Schools support PhD students during their research training at Ghent University. The Center for Molecular Modeling (CMM, http://molmod.ugent.be), led by prof. Veronique Van Speybroeck, is an interfaculty research unit at the Ghent University grouping about 40 scientists from the Faculties of Science and Engineering and Architecture. The CMM performs interdisciplinary research at the crossroads between physics, chemistry and materials engineering with the aim to design molecules, materials, and processes at the nanoscale. Excellence is pursued by stimulating interactions in the research team consisting of chemists, chemical engineers, physicists, physical engineers and bioengineers as well as with the vast network of national and international partners. The CMM has a proven track record in simulating nanoporous materials, with among others 2 ERC grants for V. Van Speybroeck in the topic. Within the CMM, prof. Louis Vanduyfhuys launched a new research track which devotes to a thermodynamic characterization of the properties of MOFs and related nanoporous materials. The group published 134 peer-reviewed papers in the last five years. Several papers were published in the highest ranked journals such as Science, Chem. Soc. Rev., Ang. Chem., JACS, Nature Mat., Nature Chem., Nat. Comm., Phys. Rev. Lett. The group has a vast experience in supervising PhD students and postdocs.
What can be your role as a computational scientist?
PhD student 1
We are looking for a PhD student to work on the “characterisation of adsorption and dielectric and refractive index response of MOFs and zeolites”.
Objectives:
- Accurate prediction of adsorption isotherms and selectivity of nanoporous materials towards various VOCs
- Accurate prediction of the dielectric constants and refractive index of nanoporous materials loaded with VOCs
Short Description of Work & Expected Results:
- Construction of accurate material-specific force fields (FFs) and/or machine learning potentials (MLPs) from ab initio training data for 50 of the most promising materials for each VOC as identified by CNRS-Montpellier
- Use these FFs/MLPs for the simulation of single- and multicomponent isotherms for the most promising 50 materials for each VOC identified by CNRS-Montpellier
- Calculation of the dielectric constant and refractive index response for the 10 most promising materials for each VOC identified as function of their guest (VOC/water) loading using ab initio techniques such as DFT
The CMM will be your host institution with prof. Van Speybroeck and prof. Vanduyfhuys as your supervisors. Two secondments are planned, one at CNRS Montpellier under the supervision of prof. Guillaume Maurin and one at SCM (Software for Chemistry & Materials) under the supervision of Stan van Gisbergen.
PhD student 2
CNRS-Montpellier is looking for a PhD student to work on the “high-throughput (HT) screening & rationalization of porous materials for selective VOC adsorption”.
Objectives:
- HT screening of the IZA (zeolites) and CoreMOF 2019 (MOFs) databases using Monte Carlo simulations to identify the porous materials.
- QSPR analysis of the so-created database using advanced statistical tools (ANN, etc.)
- In silico design of novel MOFs with improved VOC adsorption performances assembling the key features identified in the previous step using an automated assembly of structure building units (AASBU) approach (case of MOF).
Short Description of Work & Expected Results:
- Creation of an unprecedented database listing the adsorption performances of porous materials with respect to VOCs
- Establishment of structure-adsorption property relationships using advanced statistical tools
- Prediction of novel porous materials with improved adsorption performances
CNRS Montpellier will be your host institution with prof. Guillaume Maurin as your supervisor. Two secondments are planned, one at the CMM under the supervision of prof. Van Speybroeck and prof. Vanduyfhuys and one at SCM (Software for Chemistry & Materials) under the supervision of Stan van Gisbergen.
Candidate requirements
- We are looking for a computational scientist with a good background in atomistic molecular simulations, quantum mechanics, statistical physics and thermodynamics applied to material science.
- Experience with molecular simulation software (LAMMPS, DLPOLY, RASPA, Gaussian, VASP, CP2K, …) and coding (Python, C, …) is an advantage.
- You have a pro-active working style, the willingness to look beyond the borders of your own discipline and a strong motivation to work in a multidisciplinary team. You are highly motivated to become an independent researcher.
- You have excellent communication skills and have a strong motivation to collaborate with other researchers, within the CMM/CNRS Montpellier, the SENNET consortium and our networks.
- You have or will soon obtain a master’s degree of a university or international equivalent in the field of Chemistry, Chemical Engineering, Physics, Physical Engineering or a related field.
Benefits and salary
The successful candidates will receive an attractive salary in accordance with the MSCA regulations for Recruited Researchers. The exact (net) salary will be confirmed upon appointment and is dependent on local tax regulations and on the country correction factor (to allow for the difference in cost of living in different EU Member States). The salary includes a living allowance, a mobility allowance and a family allowance (if applicable). The guaranteed PhD funding is for 36 months (i.e. EC funding, additional funding is possible, depending on the local Supervisor, and in accordance with the regular PhD time in the country of origin). In addition to their individual scientific projects, all fellows will benefit from further continuing education, which includes internships and secondments, a variety of training modules as well as transferable skills courses and active participation in workshops and conferences.
On-line Recruitment Procedure
All applications proceed through the on-line recruitment portal on the https://sennet-project.eu/ website. Candidates apply electronically for one to maximum three positions and indicate their preference. Candidates provide all requested information including a detailed CV (Europass format obligatory) and motivation letter. During the registration, applicants will need to prove that they are eligible (cf. Recruited Researchers definition in Horizon Europe MSCA work programme 2021-2022, mobility criteria, and English language proficiency):
- Supported researchers must be doctoral candidates, i.e. not already in possession of a doctoral degree at the date of the recruitment.
- Researchers must be enrolled in a doctoral programme leading to the award of a doctoral degree in at least one EU Member State or Horizon Europe Associated Country, and for Joint Doctorates in at least two.
- Recruited researchers can be of any nationality and must comply with the following mobility rule: they must not have resided or carried out their main activity (work, studies, etc.) in the country of the recruiting beneficiary for more than 12 months in the 36 months immediately before their recruitment date.
The deadline for the on-line registration is 15 September 2022. Prior to the recruitment, videoconferencing (or in person, when possible) interviews between the Supervisors and the candidates will be organized. The final decision on who to recruit is communicated no later than October 2022. The selected researchers are to start their research as quickly as possible (ideally prior to 1 March 2023).
Applicants need to fully respect three eligibility criteria (to be demonstrated in the Europass CV):
- Conditions of international mobility of researchers:
- Researchers are required to undertake trans-national mobility (i.e. move from one country to another) when taking up the appointment.
- At the time of selection by the host organisation, researchers must not have resided or carried out their main activity (work, studies, etc.) in the country of their host organisation for more than 12 months in the 3 years immediately prior to their recruitment. Short stays, such as holidays, are not taken into account.
- English language proficiency:
- Network fellows must demonstrate that their ability to understand and express themselves in both written and spoken English is sufficiently high for them to derive the full benefit from the network training.

 n September 15th our colleague Maarten Cools-Ceuppens successfully defended his PhD. During his defence Maarten presented his work entitled ‘Incorporating long-range interactions and polarization in machine learning potentials with explicit electrons’. He was supervised by prof. dr. ir. Toon Verstraelen and prof. dr. ir. Joni Dambre.
n September 15th our colleague Maarten Cools-Ceuppens successfully defended his PhD. During his defence Maarten presented his work entitled ‘Incorporating long-range interactions and polarization in machine learning potentials with explicit electrons’. He was supervised by prof. dr. ir. Toon Verstraelen and prof. dr. ir. Joni Dambre.




 Last week on June 9th and 10th, dr. Sanggyu Chong visited the CMM to exchange ideas in small-group and one-on-one discussions with us. Dr. Chong recently started working as a postdoctoral researcher with prof. Michele Ceriotti at the Laboratory of Computational Science and Modeling (COSMO) at the École Polytechnique Fédérale de Lausanne (EPFL), focusing on machine learning potentials for MOFs and other materials. Before, he completed his PhD at KAIST (Korea Advanced Institute of Science and Technology), working on the electronic structure modelling of MOFs under the supervision of prof. Jihan Kim.
Last week on June 9th and 10th, dr. Sanggyu Chong visited the CMM to exchange ideas in small-group and one-on-one discussions with us. Dr. Chong recently started working as a postdoctoral researcher with prof. Michele Ceriotti at the Laboratory of Computational Science and Modeling (COSMO) at the École Polytechnique Fédérale de Lausanne (EPFL), focusing on machine learning potentials for MOFs and other materials. Before, he completed his PhD at KAIST (Korea Advanced Institute of Science and Technology), working on the electronic structure modelling of MOFs under the supervision of prof. Jihan Kim.




