Towards accurate prediction of diffusivities with machine learning techniques: light olefin diffusion in aluminophosphates during MTO conversion
Towards accurate prediction of diffusivities with machine learning techniques: light olefin diffusion in aluminophosphates during MTO conversion
Promotor(en): V. Van Speybroeck, L. Vanduyfhuys /28131 / Nanoporous materialsBackground and problem
The methanol-to-olefins (MTO) conversion on acid zeolite catalysts received a lot of attention during the last decades as a more sustainable alternative for the production of base chemicals. The MTO mechanism is very complex and still not fully understood up to date. Both aromatic and aliphatic hydrocarbon pool (HP) species inside the catalyst pores were found to assist in the conversion of methanol. As a result, the ultimate product distribution is determined by a variety of factors such as the stability of intermediates, catalytic reactivity and transport phenomena. [1,2] Especially, the role of hydrocarbon diffusion on the product selectivity remains unanswered. The ease of these diffusion paths relies on the pore topology and channel dimensions of the framework, but also on the occupation of the zeolite pores by HP species which may block the channel system and restrict diffusion. [3] Recently, it was shown that the presence of acid sites in zeolite H-SAPO-34 may also have a considerable effect on the diffusivities of small alkenes and alkanes. [4] The precise influence of the acid site strength and distribution on the other hand still remains a missing link.
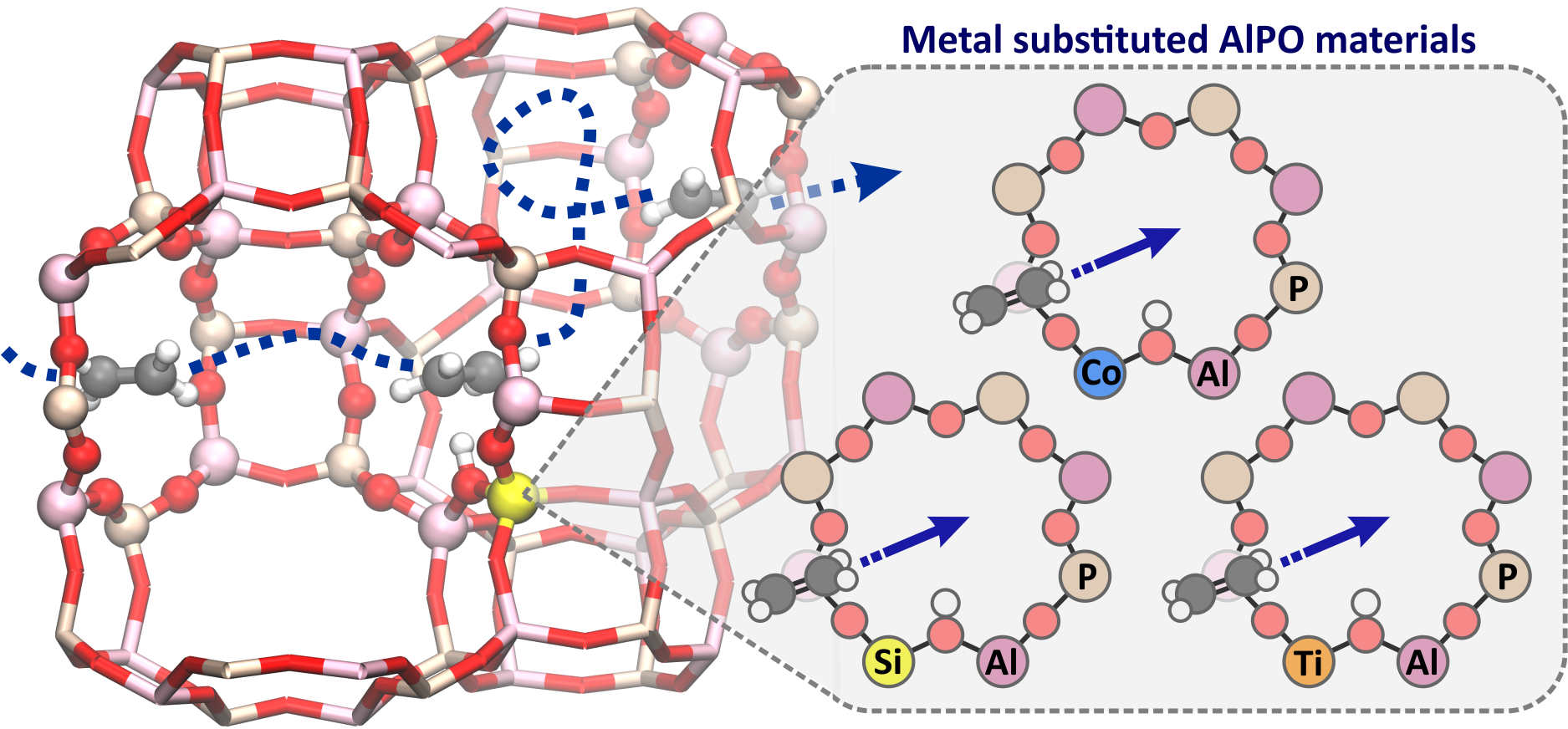
Figure 1. Schematic representation of a diffusion pathway in metal substituted aluminophosphate materials.
A proper theoretical understanding of diffusion through the zeolite micropores is mandatory for designing zeolites with a higher light olefin selectivity in the MTO process or with applications for product separation. Molecular simulations can contribute to attaining proper insight into the diffusion pathways in these materials. The diffusion phenomenon takes place at larger length and time scales and is therefore typically studied using classical force field molecular dynamics (MD) simulations. However, light olefin diffusion in a complex molecular environment can be more accurately described using a first-principle description. [4] A second complication arises because the diffusion can either occur freely or become hindered, depending on the pore topology and molecular environment. Therefore, to properly quantify hindered diffusion events, one needs to couple MD simulations with rare event sampling techniques. Performing first-principle simulations at diffusion length and time scales, though, comes at a much higher computational cost. [4] Recently, machine learning techniques have entered the computational chemistry toolbox which presents a promising alternative in this context. Ab initio data can be used to train a suitable Machine Learning Potential (MLP) that can reproduce the interatomic interactions with high accuracy at reduced computational cost, thus opening up the perspective to simulate diffusion properties at longer length and time scales and at realistic MTO operating conditions.
Goal
In the framework of this master thesis, we want to train machine learning potentials (MLPs) in order to accurately predict diffusion properties in zeolites. Furthermore, it will be investigated how the zeolite acidity can be altered to promote the diffusion of light olefins. To this end, you will study the diffusion of small hydrocarbons through the pores of metal substituted aluminophosphate zeolites in the presence of HP species which is representative for the actual MTO environment. [5] Specific case studies in which the zeolite pore system is (partially) blocked by methanol reactant and/or HP species will be taken into account. To properly model the transport phenomenon at these conditions, you will initially perform first-principle MD simulations to evaluate whether the diffusion is hindered or occurs freely. Then you will use the ab initio data to train an MLP which will be employed to model the diffusion at longer time and length scales with high accuracy. When the diffusion becomes hindered, rare event sampling techniques will be applied to quantify single event diffusion barriers. The obtained insights into the diffusion properties will be applied for constructing a model to predict diffusivities which can be compared with experimentally measured data. Ultimately, the insight into the diffusion behaviour of small hydrocarbons may assist the efforts for increasing the MTO product selectivity.
The Center for Molecular Modeling has ample experience in modeling of zeolite catalysis with advanced simulation techniques. The proposed topic is situated in a very active research field. You will be actively coached to get acquainted with the multitude of available simulation techniques. You will be involved in the running collaboration with prof. Unni Olsbye (University of Oslo) on this topic.
- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsZeolites, Diffusion, Methanol-to-olefins, machine learningReferences
[1] P. Ferri et al., ACS Catal. 9 (2019) 11542-11551.
[2] M. Gao et al., Nat. Commun. 11 (2020) 3641.
[3] Y. Shen et al. ACS Catal. 8 (2018) 11042-11053.
[4] P. Cnudde et al., Angew. Chem. Int. Ed. (2021).
[5] M. Morten et al., ChemPhysChem 19 (2018) 484-495.
Contact
Louis Vanduyfhuys
Veronique Van Speybroeck
Pieter Cnudde