Using advanced molecular dynamics simulations to unravel the drug binding into membrane proteins
Using advanced molecular dynamics simulations to unravel the drug binding into membrane proteins
Promotor(en): A. R. Mehdipour, L. Vanduyfhuys /28187 / Chemistry & BiochemistryBackground and problem
Most drugs target membrane proteins accessible on the surface of the cells. The main drug-binding sites of membrane proteins are situated in central binding sites formed within their transmembrane-spanning domains. However, growing number of drugs target lipid-facing binding sites outside of the transmembrane regions or reach into a deeper pocket using a fenestration pathway, which is accessible through membrane pathways [1] (Figure 1). Unlike a conventional ligand binding process, such drugs have a two-step binding process, including partitioning of the drug into the membrane and the subsequent binding to the binding site. The lack of a detailed picture of this binding process comes from the absence of suitable models depicting protein-lipid interaction. Therefore, realistic models of membrane proteins in their native-like environment will not only provide useful insights into the physiological role of membrane proteins, but also will enhance our understanding of drug binding in the transmembrane region [2] (Figure 1). Such models will also pave the pathway for designing new drugs with improved kinetics and efficacy. Furthermore, they can also be applied to estimate the free energy of binding of drugs into membrane proteins using realistic membranes and advanced molecular dynamics simulation methods based on methodologies from statistical physics and thermodynamics.
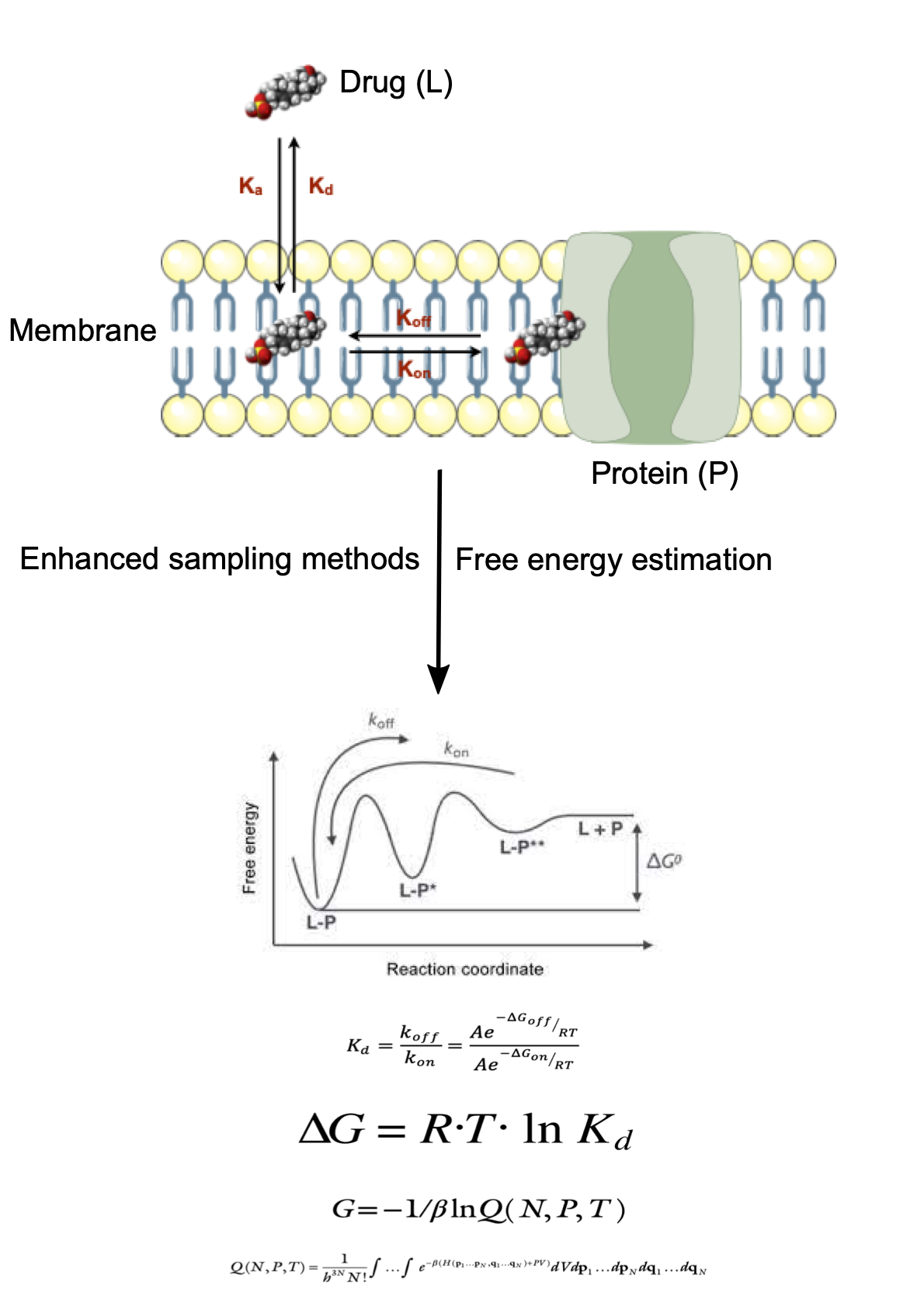
Figure 1: A schematic representation of the molecular mechanism of drug binding into a membrane-faced binding site in a two-step process.
Goal
The primary aim of this project is to simulate a therapeutically important protein (the calcium channel which is important in controlling blood pressure) in a realistic membrane (a membrane resembling human cell membrane) using the state-of-the art molecular modeling. These models will be used to look at the process of drug binding into the lipid-facing binding sites outside of the transmembrane regions or reaching into deeper pockets using fenestration pathways.
First, we use the structures of membrane proteins of interest (Ca2+ channels) bound to the drug (Nifedipine a cardiovascular drug) [3] and build a simulation setup mimicking the physiological condition with a membrane bilayer, ions, and water molecules. Initially, the student will perform equilibrium molecular dynamics simulations to check the stability and dynamics of the drug at the binding site. In the later stage of the project, the focus of the project is to calculate free energy of binding for drug using enhanced sampling methods (Umbrella sampling and Metadynamics). This will allow us to have a comprehensive understanding of how the membrane affects the binding affinity of drugs to the membrane proteins and use this information for optimal design of a novel set of drugs.
The student will model the protein in the membrane using CHARMM-GUI (CHARMM-GUI is a sever-based modeling package for building the MD simulation systems). The next step MD simulations, mainly the enhanced sampling, will be performed in an open-source MD-engine GROMACS and using PLUMED (PLUMED is an open-source library that provides a wide range of enhanced-sampling algorithms and free-energy methods). Finally, the student will construct the free energy profile based on methodologies from statistical physics/thermodynamics using PLUMED and ThermoLIB packages (ThermoLIB is a Python-based library to construct free energy surfaces (FES) as a function of CVs from the output of molecular simulations).
- Study programmeMaster of Science in Biomedical Engineering [EMBIEN]KeywordsMembrane protein complexes, drug binding, molecular dynamics (MD) simulations, Free energy calculations, statistical physics/thermodynamics.References
[1] Mol Pharmocol. 2019, 96, 527-541.
[2] PNAS, 2016, 113, 8568-8570.
[3] Cell, 2019, 177, 1495-1506.
Contact
Ahmad Reza Mehdipour