Unraveling reaction mechanisms of photocatalyzed CO2 reduction in covalent-organic frameworks
Unraveling reaction mechanisms of photocatalyzed CO2 reduction in covalent-organic frameworks
Promotor(en): V. Van Speybroeck, P. Van der Voort /28327 / Nanoporous materialsBackground and problem
Solar energy is the most abundant and inexhaustible resource on our planet. Hence, harvesting sunlight for energy production and chemical transformations will be invaluable for establishing a sustainable industrial society. Photocatalyzed CO2 reduction reactions that transform undesirable greenhouse gases into valuable chemical commodities are of great interest to further remediate CO2 emissions [1]. The conversion of light to chemical energy is accomplished by photoactive materials. Photoactive materials can promote the conversion of light by adsorbing discrete wavelengths according to the energetic difference between their occupied and unoccupied electronic bands and separating the resulting electron and hole (i.e., exciton) to either generate an electronic current or reduce and oxidize a substrate, respectively. The efficiency of photoactive materials and their catalysis of specific reactions depends on 1) the bandgap of the material, 2) the separation and migration of the generated exciton, and 3) the energetic relationship between valence and conduction bands of the photocatalyst and the potential of the targeted reaction.
To catalyze CO2 reduction at one electrode, H2O oxidation proceeds at the other electrode via photogenerated electrons and holes, respectively (Figure 1).
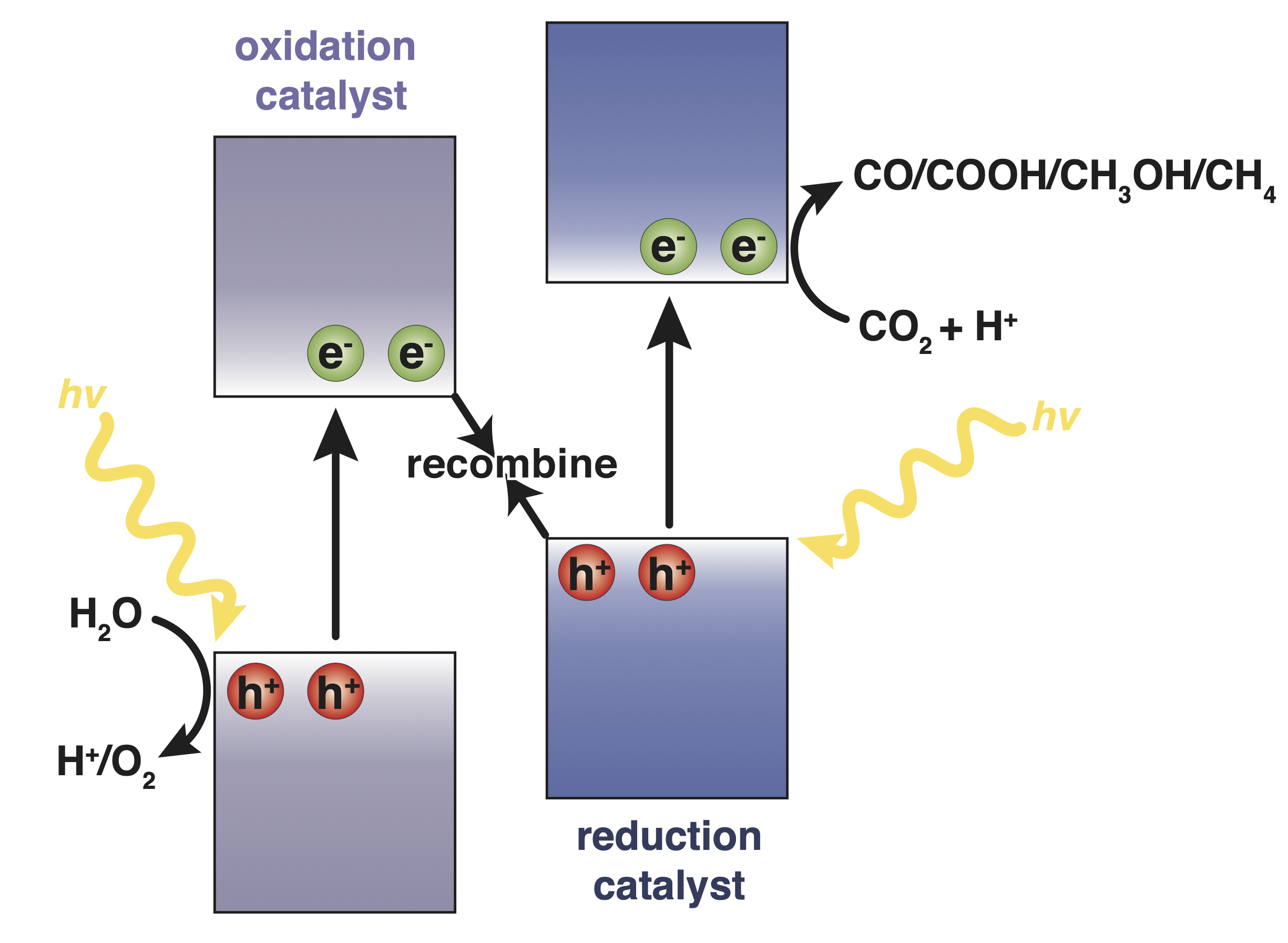
Figure 1: Illustration of Z-scheme band alignment for a photocatalytic device designed for the chemical upgrading of CO2 gas where water is oxidized at one catalyst and CO2 is reduced at the other.
Photocatalytic efficiency can be improved by the formation of heterojunctions between oxidative and reductive catalysts to promote the separation of electrons and holes. In a Z-scheme, the interface of this heterojunction promotes the recombination of low energy electrons in the oxidative catalyst, and high energy holes in the reductive catalyst while preserving spatially separated holes and electrons with superior redox activity (see Figure 1) [2]. The need for specific control of frontier band energies to design such interfaces can be satisfied by covalent organic frameworks (COFs), which are extended materials synthesized by the formation of strong covalent bonds between distinct molecular building blocks [3]. Utilizing COFs formed through π-stacking of conjugated 2D sheets with carefully selected building blocks, porous Z-scheme systems can be designed that facilitate both efficient charge transport through the layered 2D COFs and reactant adsorption for good photocatalytic efficiency.
However, the specific design of Z-scheme systems for the chemical upgrading of CO2 is additionally complicated by its chemical inertness and the inherent competition between a variety of possible product states [4]. To increase catalyst activity and skew product selectivity towards the desired reduction product, photocatalysts that encourage specific adsorbate binding modes of reactive intermediates are highly beneficial. Given that CO2 reduction is a multiproton and multielectron process with several possible products, controlling the specificity of valorization processes requires intelligent catalyst design to favor beneficial adsorption modes of all key intermediates. With the virtually unlimited modularity of COF building blocks at the molecular level in terms of structure and heteroatom inclusion, computational protocols to identify key reaction steps and adsorption modes will be invaluable for future catalyst design [5].
Goal
The goal of this project is to elucidate mechanistic details of CO2 reduction pathways in COF photocatalysts used for Z-scheme systems. The student will utilize ab-initio modeling techniques to assess the electronic structure of COF systems, including the production of electronic band diagrams and corresponding density of states plots to assess band edge alignment with respect to water oxidation and CO2 reduction potentials. They will also study the site dependent impact of COF structure and composition on molecular adsorption modes of products, reactants, and intermediates to generate reaction coordinate diagrams for CO2 transformations. By comparing the relative energetics of competing reaction pathways, the student will identify likely product trajectories. This work will supplement experimental investigations by providing information on how different catalytic active sites direct the product distribution of CO2 conversion reactions to guide future catalyst development for improved selectivity. The student will receive active coaching in the computational techniques required to study reaction mechanisms in the proposed systems and will have direct involvement with an ongoing collaboration with our experimental partners who are leading experts in the area. If the student is interested in performing part of the experiments themselves, they will have the opportunity to be trained and perform experiments in the COMOC group of Prof. Van Der Voort, who is an expert on both COF synthesis and their exploration as potential photocatalysts.
- Study programmeMaster of Science in Chemical Engineering [EMCHEM]Keywordsheterogeneous photocatalysis, covalent-organic frameworks, Z-scheme, CO2 reductionReferences
[1] Angew. Chem. Int. Ed. 2020, 59, 22894-22915.
[2] Mater. Today 2018, 21, 1042-1063.
[3] J. Am. Chem. Soc. 2020, 142, 20107-20116.
[4] Mater. Today, 2020, 32, 222-243.
[5] (a) ACS Catal. 2020, 10, 14984-15007. (b) J. Phys. Chem. C 2021, 125, 23133-23141.
Contact
Veronique Van Speybroeck