Unraveling the meaning of time in coarse-grained molecular dynamics simulations to study diffusion in heterogeneously disordered MOFs
Unraveling the meaning of time in coarse-grained molecular dynamics simulations to study diffusion in heterogeneously disordered MOFs
Promotor(en): V. Van Speybroeck /19MODEV11 / Model and software developmentIn the past decade, the advance of high-performance computing facilities and algorithms has hugely boosted the design of novel materials for a wide variety of applications. The Center for Molecular Modeling (CMM) is at the forefront of this new research field, focusing on the development of accurate molecular-level models to characterize the complex physical and chemical phenomena that take place in nanoporous materials such as zeolites, metal-organic frameworks (MOFs), and covalent organic frameworks (COFs). These largely ordered materials continue to attract great interest given their structural porosity and large tunability in terms of chemical and physical properties that is only seldom displayed in more conventional materials. From a modelling perspective, MOFs are to a large extent still treated as perfectly ordered systems, such that a perfect MOF crystal can be easily modelled by periodically repeating much smaller subunits. In recent years, however, developments in spectroscopic techniques have demonstrated that MOFs are not perfect crystals, but inherently exhibit spatial heterogeneity in various forms, such as isolated defects and disordered domains (see Figure 1), which strongly impacts the MOF’s performance [1]. To understand the behavior of these spatially disordered MOFs and bring them to functional devices, it is therefore crucial to understand on a fundamental level how this spatial disorder impacts the material’s properties under real-life conditions.
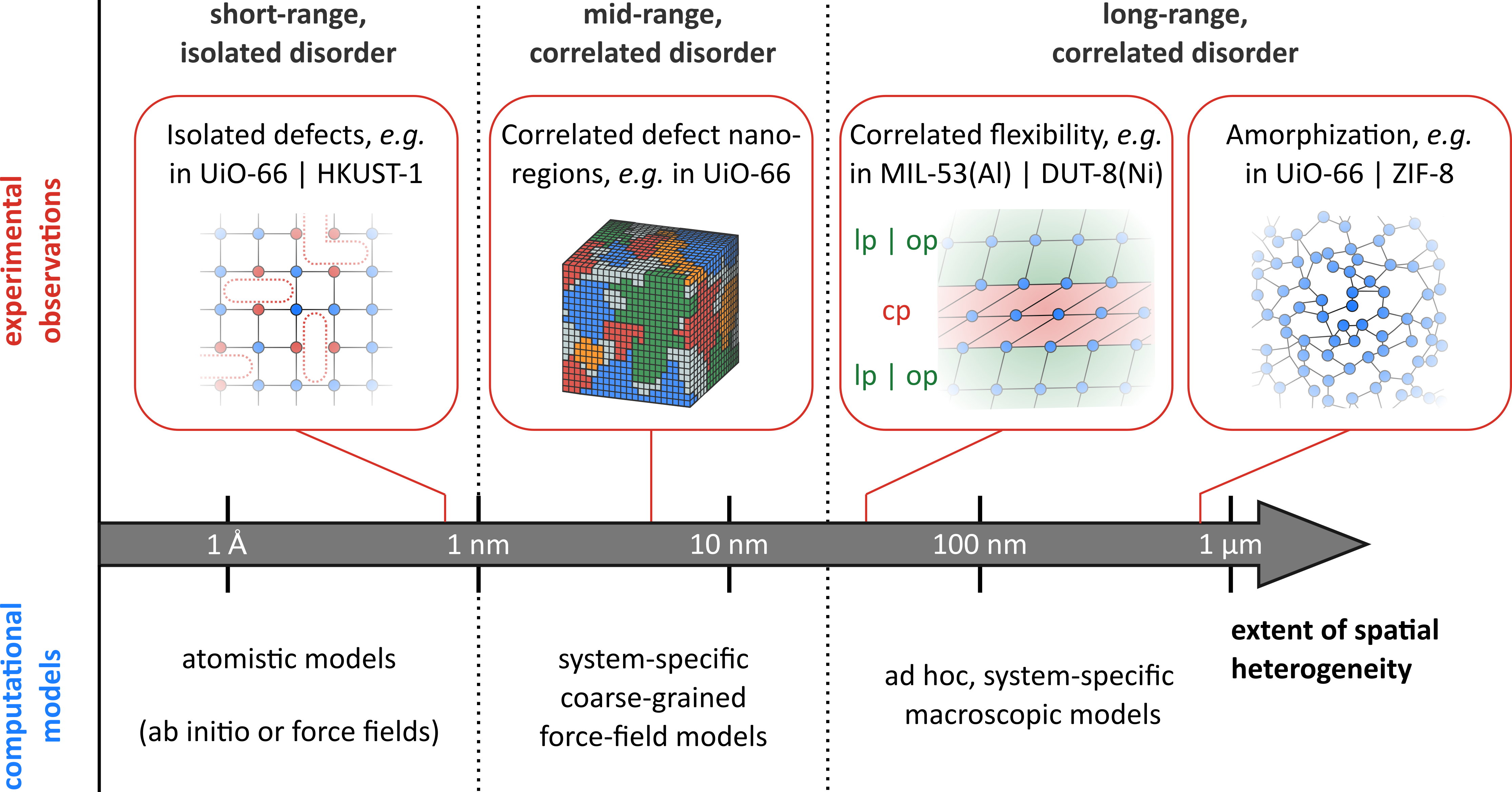
One of the crucial challenges in the computational modelling of nanoporous materials therefore consists of bridging the gap between the length scale that is accessible in current computer simulations – corresponding to the aforementioned subunits typically a few nanometers in diameter – and the size of actual MOF crystals, which might grow to several micrometers. While simulations performed on these nanosized models may already provide important insights into the properties exhibited by MOFs, they necessarily neglect the rich variety of spatial disorder that is intrinsically present in these materials but extend over longer length scales, as indicated in Figure 1. One method to bridge the gap between these computational and experimental length scales are coarse-grained (CG) models, in which atoms are no longer treated individually but instead collected in beads. These CG techniques are often used in biomolecular systems like proteins, where a huge number of atoms need to be treated. While multiple CG techniques have already been developed for these biomolecular systems, such as the MARTINI force field [2], CG force fields for nanoporous materials are still very limited [3].
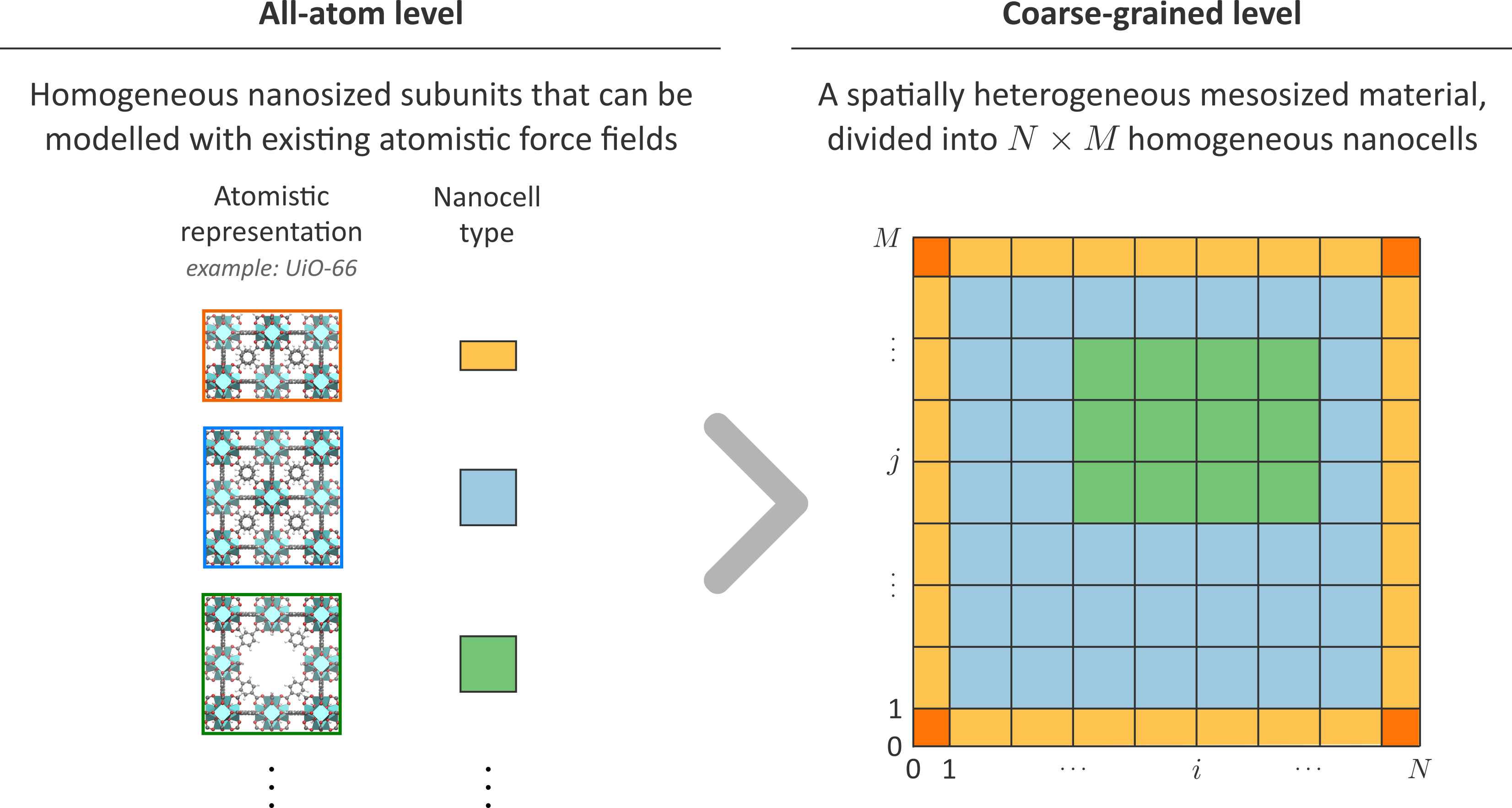
Recently, we implemented a coarse-graining method based on the variational minimization of the Kullback-Leibler divergence, an alternative and widely-used technique that has not yet been systematically adopted for MOFs [4]. One of the major advantages of this alternative technique is that it allows one to hierarchically build on atomistic force fields, which were already thoroughly validated with experiment. Therefore, this technique is ideally suited to systematically increase the length scale accessible in molecular simulations. This idea is visualized in Figure 2, in which the right-hand side shows a spatially disordered MOF crystal containing various domains, which themselves are composed of different homogeneous subunits (the NxM cells). For each of the nanosized subunits corresponding to a different type of disorder, a CG model can be derived using the variational minimization of the Kullback-Leibler divergence, building further upon the all-atom force field description indicated in the left-hand side of Figure 2. As these all-atom force fields are already a simplified version of the quantum mechanical potential energy surface, the CG models derived here may be considered a logical extension to further increase the time and length scale in the description of spatially disordered nanoporous materials.
Goal
Within this thesis, we will specifically investigate the meaning of time within a coarse-grained molecular dynamics simulation of the nanoporous MOF UiO-66. As CG techniques map different atoms into one CG bead, they also neglect the often high-frequency motions of the atoms inside a given bead. In this sense, the time evolution of the coarser system is disrupted. The advantage of this artificial acceleration of time is that more relevant configurations can be sampled in only a fraction of the computer time necessary to sample the corresponding atomistic configurations. This speed-up is often an attractive feature, as it allows to investigate phenomena that not only take place on longer length scales, but also on longer time scales. However, this coarse-graining of time in molecular simulations is only poorly understood, and dynamic phenomena are therefore typically beyond the scope of conventional force fields such as MARTINI [2]. While one can resort to approximate techniques, such as the introduction of an ad hoc speed-up factor to correct for the expected dynamics of the system [5], the applicability of this rule of thumb is necessarily limited and not based on physical insights.
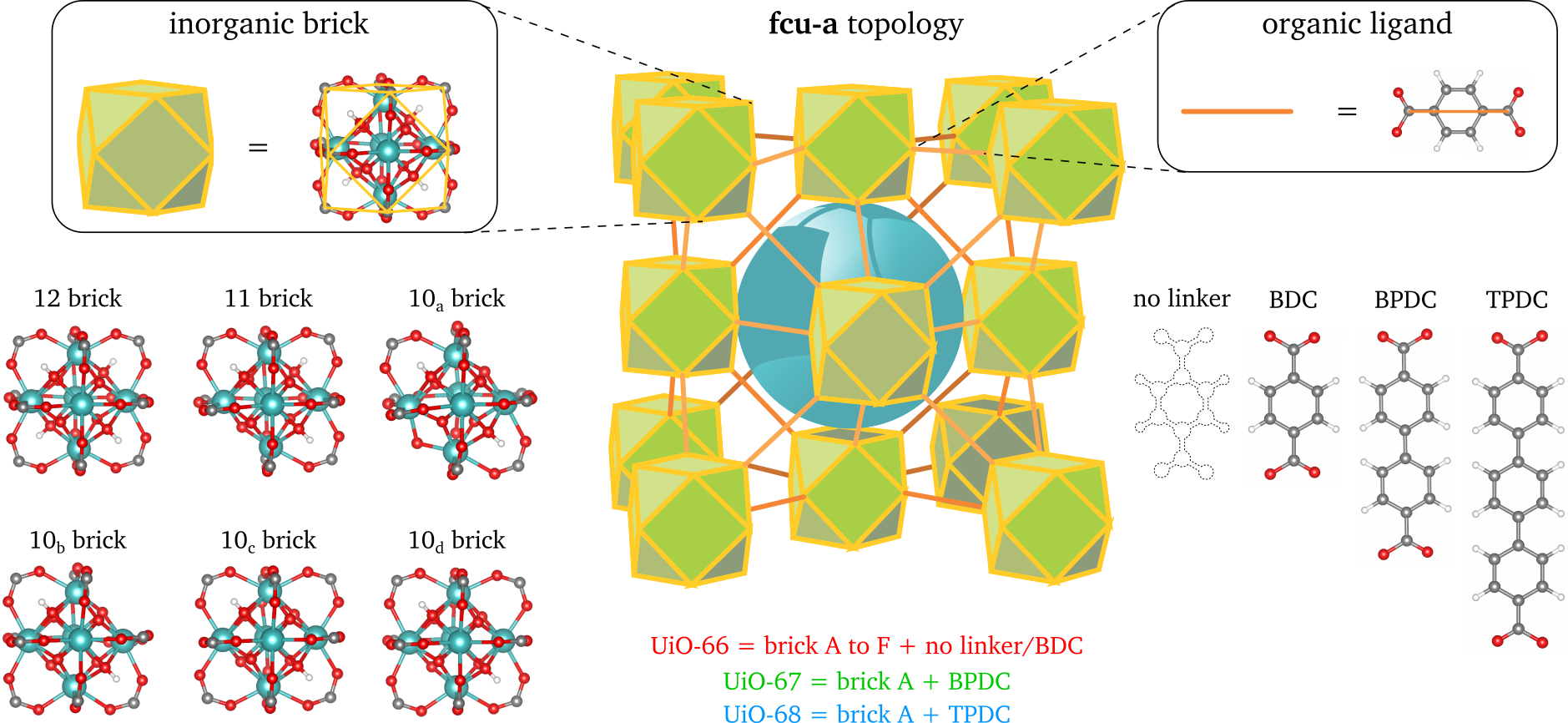
To thoroughly investigate the concept of time within coarse grained molecular dynamics simulations, we will consider the industrially relevant case of methane and carbon dioxide diffusion in UiO-66. For this rigid MOF, both in-house atomistic data and experimental data on the diffusion of these molecules are available [6]. The UiO-66 material is a showcase example where different types of disorder are present, as indicated in Figure 3, so that accurately modelling this material requires the introduction of larger models (right-hand side of Figure 2) [7]. As the diffusion of guest species is typically a kinetic process, a proper description of the time evolution of the system is required. Within this thesis, it is the aim to systematically study how methane and carbon dioxide diffuse through atomistic and different CG models of this material. Both defect-free and defective atomistic models will be studied and compared with the coarse grained model. As such, insight into the different factors influencing the speed-up factor in coarse-graining will be obtained. Inspired by a similar study on CG diffusion in pure solvents [8], the aim of this thesis is therefore to propose a systematic approach to define time in CG simulations for nanoporous materials. If successful, these results can be further extended to explore time for other dynamic phenomena in MOFs, such as phase transitions and rate constants.
The student will be actively coached to make him/her acquainted with the several advanced simulation and coarse-graining techniques early in the thesis year, and to transfer necessary programming skills needed to perform the research. A strong interest in programming is, however, a prerequisite.
Aspects
Master of Science in Engineering Physics: This thesis subject is closely related to the following clusters of elective courses: NANO and MODELING. Physics aspect: exploring the concept of time in coarse-grained models; Engineering aspect: exploration of the effect of spatial disorder on the diffusion of industrially relevant molecules in metal-organic frameworks.
- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLINGKeywordsDiffusion, time, Coarse graining, spatial disorder, hierarchical models, force fieldReferences
[1] I. L. C. Buurmans and B. M. Weckhuysen, "Heterogeneities of individual catalyst particles in space and time as monitored by spectroscopy," Nat. Chem., vol. 4, pp. 873-886, 2012.
[2] S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, "The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations," J. Phys. Chem. B, vol. 111, no. 27, pp. 7812-7824, 2007.
[3] J. P. Dürholt, R. Galvelis and R. Schmid, "Coarse graining of force fields for metal-organic frameworks," Dalton Trans., vol. 45, no. 10, pp. 4370-4379, 2016.
[4] I. Bilionis and P. S. Koutsourelakis, "Free energy computations by minimization of Kullback-Leibler divergence: An efficient adaptive biasing potential method for sparse representations," J. Comput. Phys., vol. 231, no. 9, pp. 3849-3870, 2012.
[5] T. Negami, K. Shimizu and T. Terada, “Coarse-grained molecular dynamics simulations of protein-ligand binding,” J. Comput. Chem., vol. 35, no. 25, pp. 1835-1845, 2014.
[6] Q. Yang, H. Jobic, F. Salles, D. Kolokolov, V. Guillerm, C. Serre and G. Maurin, “Probing the Dynamics of CO2 and CH4 within the Porous Zirconium Terephthalate UiO‐66(Zr): A Synergic Combination of Neutron Scattering Measurements and Molecular Simulations,” Chem. Eur. J., vol. 17, no. 32, pp. 8882-8889, 2011.
[7] S. M. J. Rogge, J. Wieme, L. Vanduyfhuys, S. Vandenbrande, G. Maurin, T. Verstraelen, M. Waroquier and V. Van Speybroeck, "Thermodynamic Insight in the High-Pressure Behavior of UiO-66: Effect of Linker Defects and Linker Expansion," Chem. Mater., vol. 28, no. 16, pp. 5721-5732, 2016.
[8] J. G. E. M. Fraaije, J. van Male, P. Becherer and R. Serral Gracià, “Calculation of Diffusion Coefficients through Coarse-Grained Simulations Using the Automated-Fragmentation-Parametrization Method and the Recovery of Wilke–Chang Statistical Correlation,” J. Chem. Theory Comput., vol. 14, no. 2, pp. 479-485, 2018.
Contact
Veronique Van Speybroeck