Tuning the optical properties of ruthenium photocatalysts for efficient light capture in covalent organic frameworks
Tuning the optical properties of ruthenium photocatalysts for efficient light capture in covalent organic frameworks
Promotor(en): V. Van Speybroeck /17SPEC03 / SpectroscopyCovalent organic frameworks (COFs) are a very recent class of nanoporous, crystalline materials. They possess an enormous potential as materials for energy storage, gas capture, and heterogeneous catalysis [1]. COFs are composed solely of organic building blocks, interacting via strong covalent bonds. By varying the composition of these building blocks, a huge versatility of COFs can be envisaged. This principle paves the way to engineer these materials on a molecular scale, for instance by increasing the length of the building block to synthesize materials with larger channels, or by adding nitrogen moieties to allow for specific supramolecular interactions (see Figure 1). These nitrogen moieties, for instance present in the bipyridine ligand of Figure 1 (top right), act as a docking site to which different molecular complexes can be anchored, such as the ruthenium complex in Figure 1.
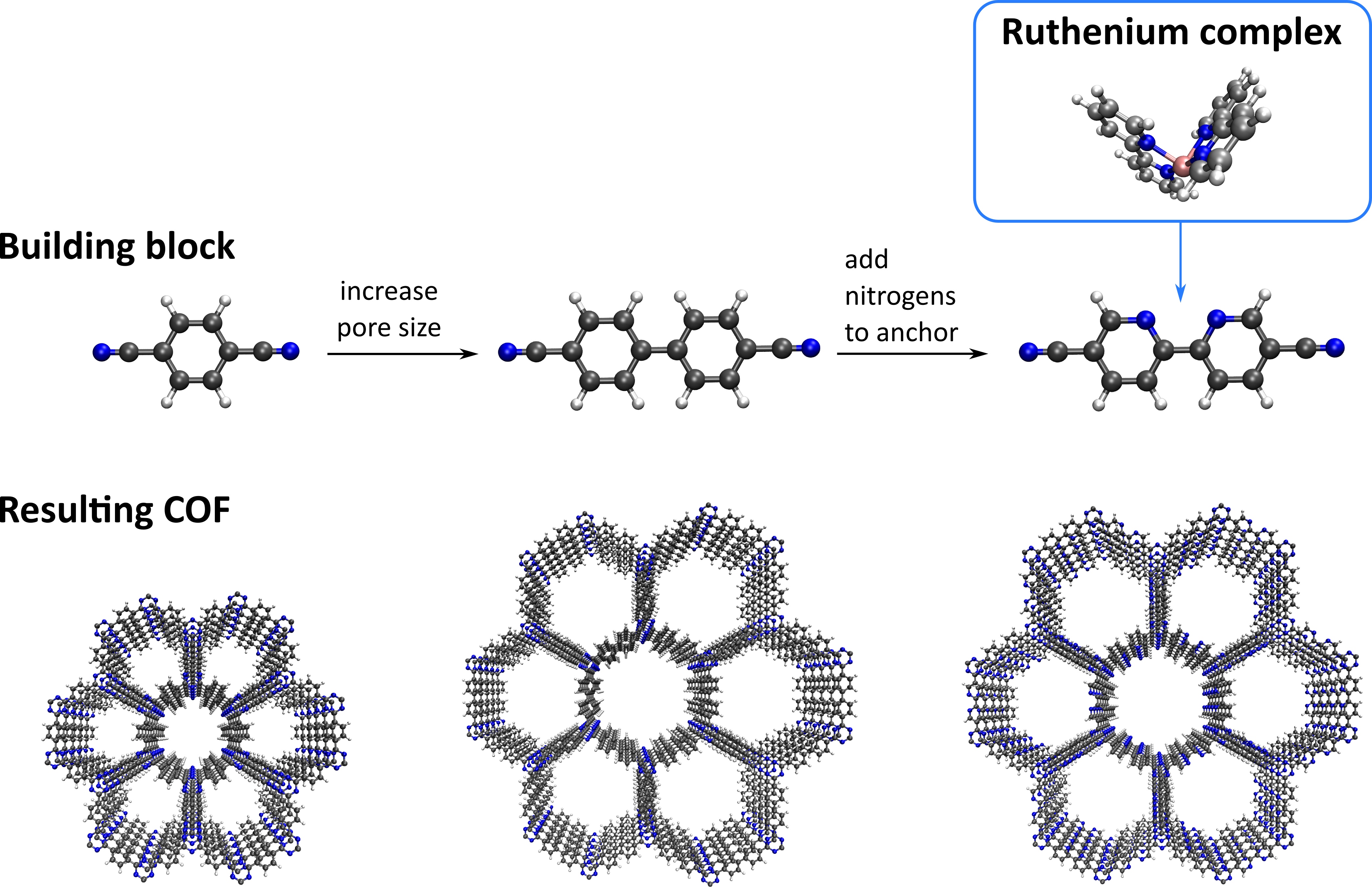
Anchoring these molecular complexes in a framework material offers several advantages compared to using these complexes in a solution. First, thanks to the creation of favorable docking sites – the bipyridine linker – the molecular complexes will preferentially anchor on these sites, and hence be spatially separated. This ensures that the different molecular complexes in the framework do not aggregate or interfere with each other. Second, these anchored complexes can be extracted from the framework material if necessary, and are hence not lost after use. For these reasons, catalytically active complexes, i.e. complexes that accelerate a given reaction without being consumed appreciably, are often anchored in these materials to form a so-called heterogeneous catalyst. Compared to their homogeneous analogue, the aforementioned advantages result in a higher yield and lower waste, driving a sustainable or green approach to chemistry. Especially of interest are photocatalysts such as ruthenium: molecular complexes which are excited upon the absorption of light, and which are typically very expensive. However, while anchoring these expensive complexes in the COF material leads to clear advantages, their photocatalytic properties can be altered [2]. Specifically, changing the ligand results in changes in the electronic structure of the catalyst, which needs to be thoroughly investigated on a molecular level in order to identify the optimal ligands that need to be used in synthesis.
Goal
This thesis aims to investigate the influence of the used COF components on the optical properties of the ruthenium complex by means of a quantum mechanical description of relevant excited states. In this respect, time-dependent density-functional theory (TD-DFT) is a powerful computational technique that allows to study the low-lying excited states of molecular complexes. Current wavefunction-based methodologies are still computationally very expensive, impeding the study of larger systems or systems containing transition metal atoms. TD-DFT in its linear response formulation [3], however, allows for the accurate prediction of vertical electronic excitation energies at a reasonable cost. Moreover, it can be performed in combination with molecular dynamics calculations or with solvated models, resulting in good agreement with experimental results. Ruthenium complexes in homogeneous phase have been studied using TD-DFT to understand the electronic structure of the excited states, to investigate the influence of different ligands or functional groups and to study the metal-to-ligand charge transfer (MLCT) processes (see Figure 2). Nevertheless, it is still unclear how the optical properties of the ruthenium complex are altered when coordinated to ligands relevant in the synthesis of COFs.
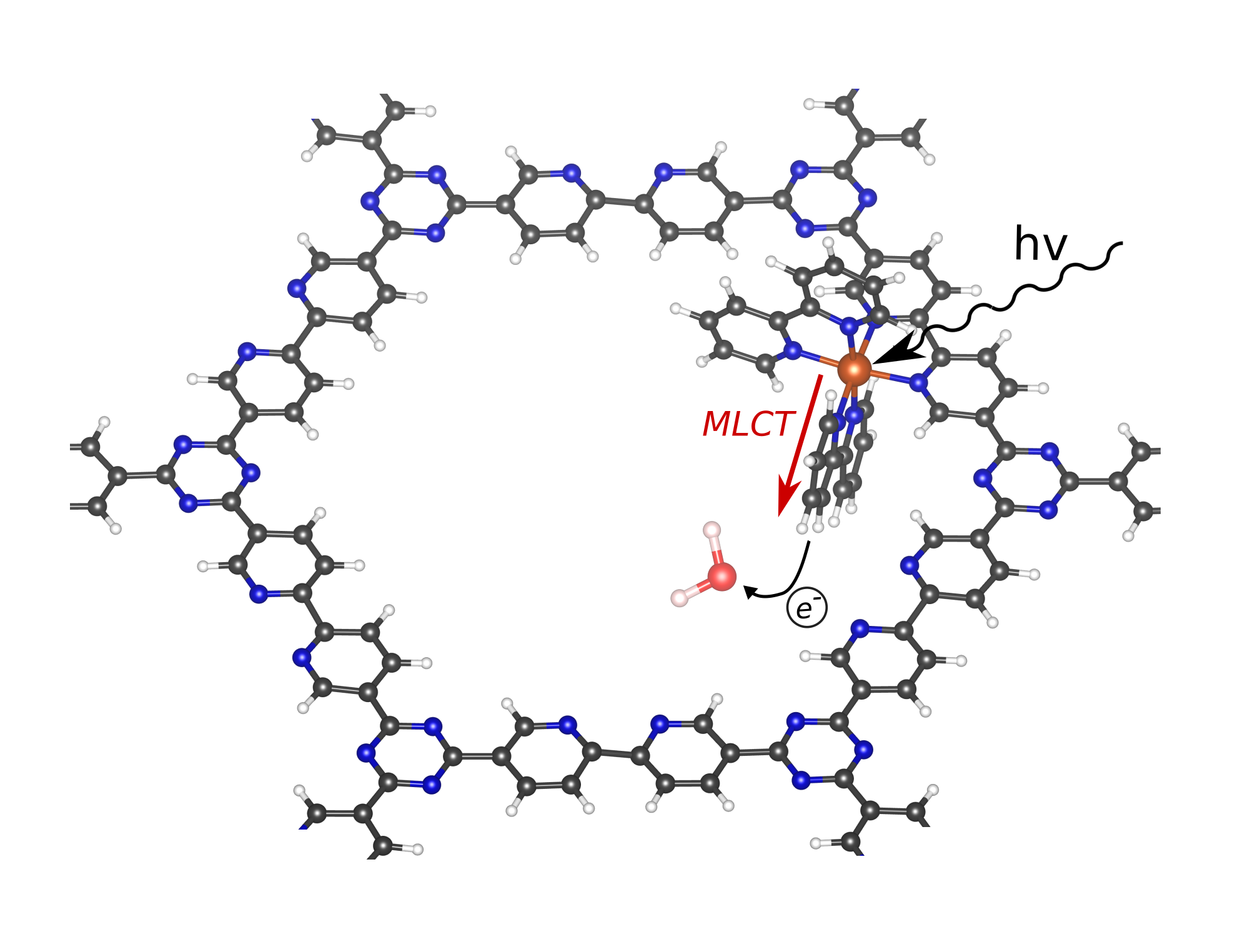
In this project, you will use TD-DFT to investigate such complexes relevant for COFs. A screening study of a range of experimentally available ligands will be performed to understand in detail the influence of the complexated transition metal on light absorption and MLCT properties. These results will then be compared with band structure calculations on the periodic materials in order to fully understand the influence on the photocatalytic properties of the complexes. With this input, the experimentalists can choose which ligands need to be taken into consideration to rationally construct new COFs.
Aspects
Physics aspect: Modelling interaction between photocatalytic molecular complexes and framework materials.
Engineering aspect: Computationally identifying the best ligand material to be used to anchor these complexes to be used in sustainable chemistry.


- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) NANO, MODELLINGKeywordslight-induced electronic transitions, Heterogeneous Catalysis, photocatalysis, time-dependent density-functional theory, metal-to-ligand charge transfer, Covalent organic framework
Contact
Veronique Van Speybroeck