Tuning catalytic activity in methanol-to-hydrocarbon conversion with aluminum distribution
Tuning catalytic activity in methanol-to-hydrocarbon conversion with aluminum distribution
Promotor(en): V. Van Speybroeck /28322 / Nanoporous materialsBackground and problem
Zeolites are crystalline aluminosilicates that can be used for a broad range of applications such as gas separation and capture and as highly efficient catalysts. Their structure consists of tetrahedrally coordinated SiO4/AlO4 units which form a network of pores and channels. Each aluminum tetrahedron is charge-balanced by a counterion; in acid zeolites this manifests as one protonated oxygen per AlO4 unit. These protons are strong Brønsted acid sites (BAS) that act as catalytic centers in the material. For methanol-to-hydrocarbon (MTH) processes, BAS content is tuned to balance activity and selectivity [1]. Higher Al content correlates with higher activity/methanol conversion because of the increased number of active sites. However, close proximity between these sites, and their adsorbed intermediates, may promote side-reactions and secondary transformations of primary products thereby diminishing catalytic specificity. Computational methods will be indispensable for assessing the explicit role of Al-distribution in governing reaction pathways, product selectivity, and overall catalyst activity. Additionally, simulated spectroscopic signals can be employed to evaluate the success of experimental methodologies for controlling Al-siting and distribution [2, 3].
There is clear experimental evidence that the aluminum distribution has a distinct influence on the catalytic and transport properties in zeolites [4]. We recently found that the transport of alkenes formed in the MTH process is severely affected by the Al distribution [5]. Product distribution also shows a strong dependence on acid site density, where close proximity between active sites promotes aromatic reactivity, while isolated active sites favor propene formation [6]. Yet, from a theoretical point of view, reactions in zeolites tend to be studied by selecting one—and only one—specific location of the Al atom in the zeolite unit cell. This obviously creates a gap between theory and experiment, as the true material likely hosts Al at multiple sites. Therefore, to generate more realistic simulations of the MTH process at operating conditions, it is important to consider multiple Al sites per unit cell. Al-distribution and siting is commonly probed through nuclear magnetic resonance (NMR) spectroscopy, where simulated signals can be used to validate the relative position of Al substitutions in structural models.
Computational techniques to calculate theoretical NMR properties enable systematic investigations to deduce the precise effect of local structural properties and aluminum location on the chemical shifts observed in complex materials such as zeolites. Indeed, the locations of distinct BAS have been characterized through this combination of theoretical and experimental NMR spectroscopy [7]. However, the presence of guest species such as water can have a strong influence on NMR signals and chemical reactivity, which reveals the need for operando modeling. Differently from static simulations, it allows to explicitly take into account temperature effects and the mobility of the adsorbates, thereby providing results that are more in line with the experimental ones.
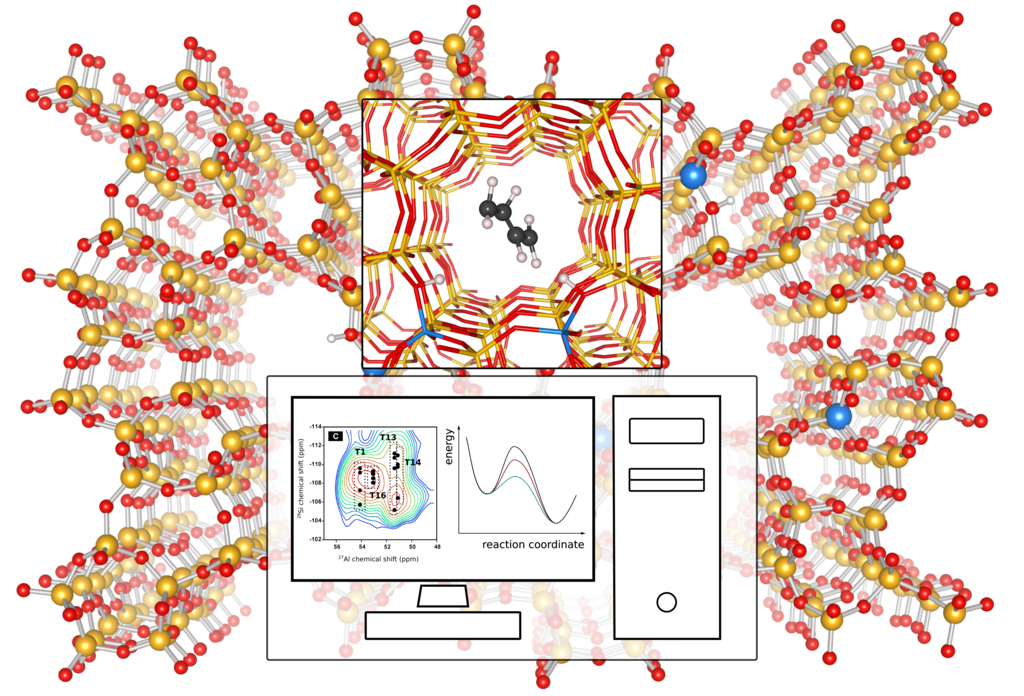
Figure 1: Molecular simulations can unravel the spatial distribution of Al sites in zeolites – through comparison with experimental NMR spectra – and determine its effect on the MTH reaction. Spectrum adapted from Ref. [7].
Goal
This thesis aims to apply state-of-the-art static and molecular dynamics simulations to model the effect of Al and BAS distribution on key steps of the catalytic cycle and deactivation process in MTH conversion.
First, accurate structural models of unit cells containing multiple Al-atoms will be generated. Herein we will not only focus on some important industrially relevant zeolites such as ZSM-5, SAPO-34, SSZ-13 but also on some zeolites which are more easily accessible for experimental benchmark purposes having a one-dimensional architecture (TON, AFI,…). For each of these zeolites, the student will generate a library of possible structures for various Si/Al ratios that will provide a database of NMR signals for experimental validation. The main focus of the project is to then calculate activation energies of some key steps in the active MTH mechanism/deactivation process with different Al-pair orientations to observe differences in transition-state-directed selectivity. Furthermore, the influence of water on the BAS and on the reaction mechanism will be touched upon, by explicit consideration of the presence of water. This will allow us to understand how Al-distributions affect the catalytic outcome of an industrially relevant reaction and pave the way towards computationally-driven catalyst engineering.
- Study programmeMaster of Science in Chemical Engineering [EMCHEM]KeywordsZeolites, catalysis, activation energy, Density functional theory, nanoscale modelingReferences
[1] ACS Catal. 2018, 8, 2, 770-784
[2] Ind. Eng. Chem. Res. 2018, 57, 11, 3914-3922
[3] Angew. Chem. Int. Ed. 2018, 57, 3742
[4] Catal. Rev. Sci. Eng. 2012, 54:2, 135-223
[5] Angew. Chem. Int. Ed. 2021, 60, 10016
[6] J. Am. Chem. Soc. 2019, 141, 37, 14823-14842
[7] J. Phys. Chem. Lett. 2018, 9, 19-24
Contact
Veronique Van Speybroeck